Clinical Mimicry: Fabry Disease Masquerading as Lupus?
ABSTRACT: A 36-year-old woman presented with Raynaud phenomenon, arthralgia, photosensitive rash, proteinuria, dry eyes and dry mouth, and alopecia.
There are very few cases of coexisting systemic lupus erythematosus (SLE) and Fabry disease.1 In most of those cases, patients present with signs and symptoms of SLE (fever, rash, arthralgia) with renal involvement (proteinuria, hematuria).2,3 In this article, we report on a case that reminds clinicians of the propensity of Fabry female heterozygotes to have significant renal morbidity and the idea that Fabry disease may masquerade as SLE.
Background
Fabry disease, or Anderson-Fabry disease, is a rare X-linked hereditary lysosomal storage disorder characterized by deficient enzyme activity of alpha-galactosidase A. This enzyme is a lysosomal hydroxylase that when deficient causes an abnormal systemic accumulation of the glycosphingolipid ceramide trihexoside in various tissue and organs.4 The deposition of ceramide trihexoside in lysosomes is found predominantly in vascular endothelial cells, smooth muscle cells, cardiac myocytes, leukocytes, fibroblasts, and epithelial cells of ganglion, corneal, and renal origins.
Fabry disease is a sex-linked disorder that is carried on the Xq22 chromosome; the incidence in men is 1 case in 40,000 persons. In most men who are carriers, or hemizygotes, the disease develops with severe manifestations beginning in childhood or adolescence. They include corneal, GI, cardiomyopathy and arrhythmia, CNS (stroke), peripheral nervous system (acroparesthesias), skin (angiokeratomas), and renal (hematuria, proteinuria, and renal failure) involvement, usually by the fourth decade. The disease traditionally has been characterized by death, typically resulting from end-stage renal disease (ESRD); complications may occur during the fifth decade.5-7
Traditionally, heterozygote women had disease manifested in a milder form because of an intermediate level of alpha-galactosidase A enzyme activity6,7; however, more recent literature points to the opposite effect. A review of cases of women with heterozygous Fabry disease found that these patients are not just carriers but that they have a significant burden of disease and impaired quality of life.8 Other articles have cited more and more cases showing that many women are indeed symptomatic at presentation and may have severe disease complications.9-11
Symptoms such as those seen in this patient are somewhat common. A presentation of Fabry disease may be more common than was previously thought, which has a greater impact on the approach to and diagnosis of this disease in the clinical setting.8,10,11 The clinical characteristics that manifest in women range from fever, myalgia, and generalized weakness to whorl-like corneal epithelial dystrophy, angiokeratomas, and acroparesthesias. Up to 20% of women will experience major cerebrovascular, cardiac, or renal events, at a median age of 46 years.9
Case presentation
The patient, a 36-year-old white woman, received a diagnosis of isolated Raynaud phenomenon and was treated for it in 2003. Three years later, after continued follow-up and successful treatment, the patient was found on random urinalysis to have isolated 2+ proteinuria. Basic chemistries, including blood urea nitrogen and creatinine levels, were unremarkable. A nephrology workup revealed nephritic-range proteinuria (greater than 1.05 g/24-h collection). Antinuclear antibody (ANA) test results were negative. The cause of proteinuria remained unclear. Proteinuria was managed with an angiotensin-converting enzyme inhibitor.
During the following few months, the patient was found to have hypothyroidism and intermittently complained of facial paresthesia; the subsequent diagnosis was trigeminal neuralgia. On follow-up, the patient complained of arthralgia, predominantly localized to the right knee. Ultrasonographic investigation showed patchy synovitis of the right knee, with no fluid.
In subsequent follow-up visits, a fine morbilliform, scattered, nonraised, nonpruritic photosensitive rash was revealed initially on the patient's lower extremities. The rash was found later on the upper extremities and remained in sun-exposed areas.
A clinical diagnosis of SLE was made in July 2008. Therapy was started with hydroxychloroquine, 200 mg bid, and nortriptyline, 20 mg qd. On follow-up for SLE over the subsequent few weeks, the patient complained of depression, anxiety, and fatigue. A selective serotonin reuptake inhibitor was added, and the patient was scheduled for a renal biopsy.
A thorough lupus workup revealed alopecia, dry mouth, and dry eyes; the result of another ANA test was positive (higher than 1:160), rheumatoid factor (RF) was 14.9 IU/mL (normal, lower than 15 IU/mL), and the ribonucleic protein (U1RNP) antibodies level was 134 AU/mL (normal, lower than 100 AU/mL). Anti-Smith, anti–double-stranded DNA, antihistone, lupus anticoagulant, Sjgren Abs, and antineutrophil cytoplasmic antibodies serology results remained negative. The patient had normal complement and C-reactive protein levels and a normal erythrocyte sedimentation rate. Her right knee remained swollen.
On the basis of her symptoms, positive ANA with weakly positive U1RNP antibodies, and the presence of persistent proteinuria, a diagnosis of SLE with coexisting depression and fibromyalgia was made. Treatment with nortriptyline was stopped, pregabalin was started, and duloxetine and hydroxychloroquine were continued. Arrangements were made for the patient to seek counseling
for her depression, which improved.

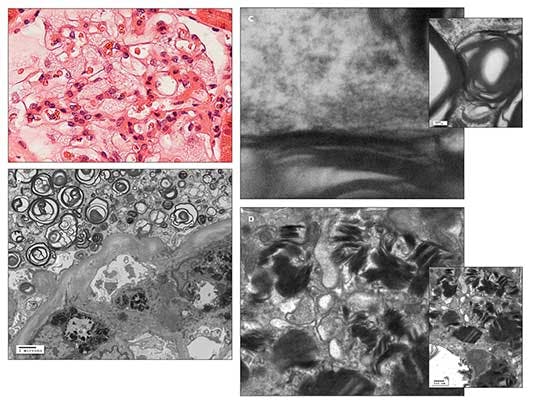
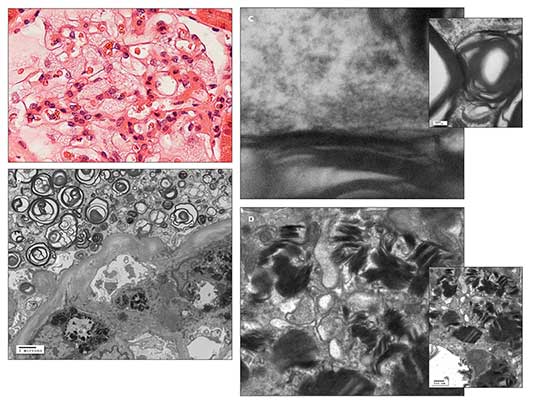
Figure – A light microscopic photomicrograph with hematoxylin and eosin staining (A) shows a glomerulus with enlarged podocytes (visceral epithelial cells), revealing a finely vacuolated “foamy” appearance. An electron microscopic photomicrograph (B) shows abundant electron-dense whorled osmiophilic lamellated myelin-like bodies in the cytoplasm of an enlarged glomerular podocyte (visceral epithelium) and lamellated inclusion bodies in the cytoplasm of the glomerular mesangial cell. An electron micrographic photomicrograph (C) shows high magnification of the periodicity (about 5 nm) of the lamellated myelin-like bodies. An electron micrographic photomicrograph (D) shows high magnification of lamellated inclusion bodies with dense layers in parallel array (“zebra body”) in the cytoplasm of the glomerular mesangial cell. These findings are suggestive of Fabry disease. (Photographs courtesy of Dr Kim.)
Light microscopic, immunofluorescence, and electron microscopic studies of the renal biopsy specimen showed no features of lupus nephritis. However, there was fine “foamy” vacuolation in the enlarged glomerular podocytes, tubular epithelium, and interstitial cells on light microscopy; electron-dense whorled osmiophilic lamellated myelin-like bodies with 5-nm periodicity in the cytoplasm of the glomerular podocytes and lamellated inclusion bodies with dense layers in parallel array (“zebra body”) in the cytoplasm of the glomerular mesangial cells on electron microscopy; and no immune-type deposits on immunofluorescence. These findings are suggestive of Fabry disease associated with focal segmental glomerulosclerosis (FSGS) and focal global glomerulosclerosis (Figure). Further alpha-galactosidase A and DNA testing confirmed the presence of Fabry disease.
Discussion
The coexistence of SLE and Fabry disease is uncommon. In this case, there are lysosomal inclusions with osmiophilic lamellated electron-dense myelin-like bodies seen on an electromyogram, suggestive of Fabry disease. However, there is no evidence of lupus nephritis. We present an interesting case of the coexistence of Fabry disease and FSGS without ESRD. In the literature, Van Loo and associates12 presented a case of a heterozygous woman with Fabry disease and ESRD who was found to have FSGS. Until now, publication on combined Fabry disease and FSGS has been limited; more research is needed.
The mechanism of antibody formation is based on the accumulation of ceramide trihexoside.1,2,13,14 It has been established that the lipids that accumulate in Fabry disease (and other similar storage diseases, such as Gaucher) are strongly immunogenic, resulting in various abnormal antibody formations.1,2,13,14 This explains the weakly positive ANA, RF, and RNP antibodies in our patient.
The immunogenic ceramide trihexoside results in antigen-antibody complex, as well as renal glomeruli deposition, leading to FSGS.1,13 The pathogenic relationship remains unclear (whether the patients were predisposed to SLE or are patients with Fabry disease that is masquerading as lupus, or the diseases are concomitant, each manifesting because of flares of the other disease); Fabry disease and SLE have several common clinical symptoms (Table). Some of the literature suggests that the chronic antigenic stimulation of the lipids, and thus the formation of autoantibodies, can lead to immune complex disorders on a scale that ranges from lupus15 to multiple sclerosis.16

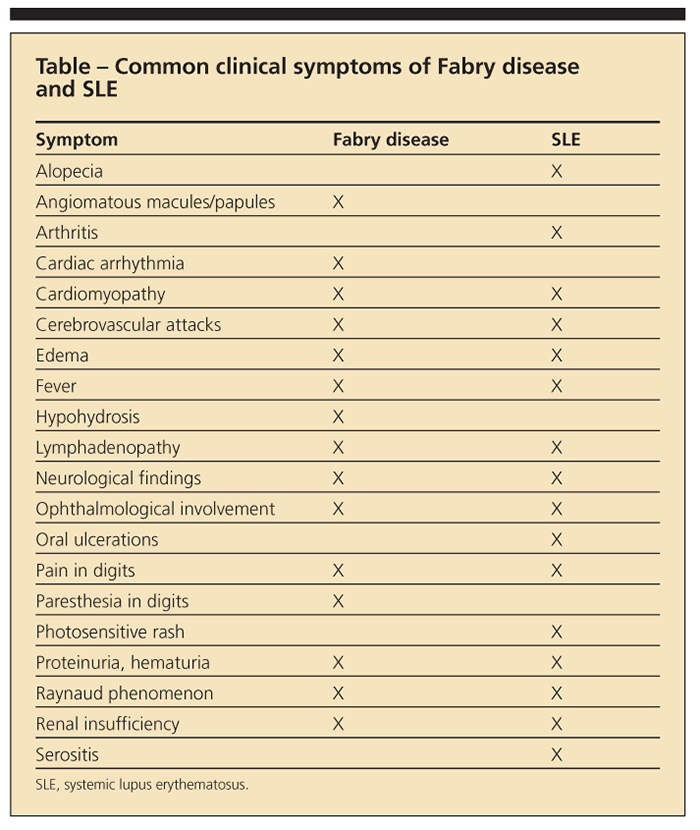
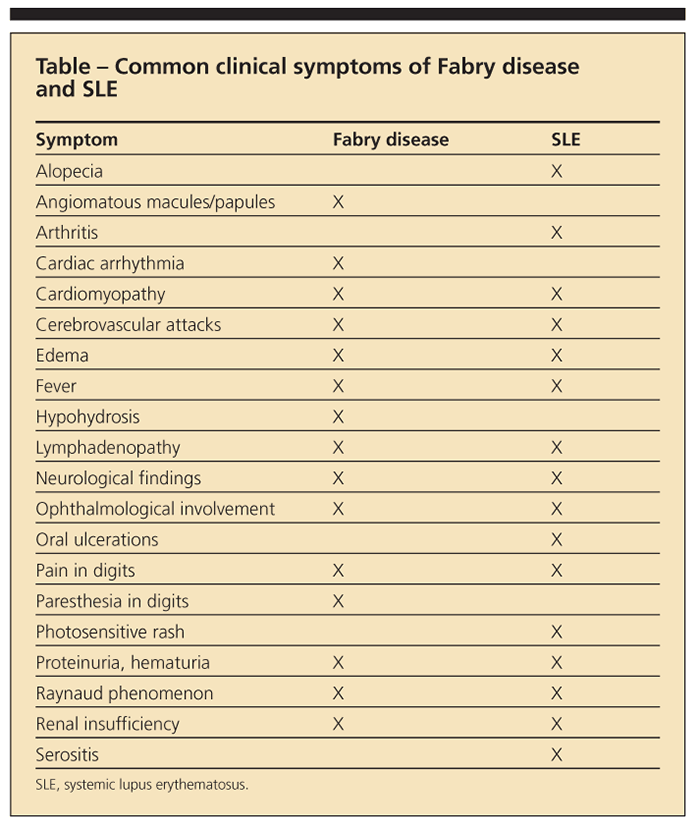
Significant renal morbidity can develop in Fabry female heterozygotes. In one study, 12% of patients with Fabry disease on hemodialysis were women17; in addition, 17 of 57 female patients (33%) with Fabry disease studied at the NIH exhibited proteinuria (at least 500 mg of protein), and at least 60% had some proteinuria on presentation.
A recent review emphatically demonstrated the significant morbidity and mortality in this subgroup of patients with Fabry disease previously viewed as having an excellent prognosis.18 In this case report, the patient's alpha-galactosidase A level was 2.1 nmol/h/mg (normal range, 28 to 80 nmol/h/mg). Genetic testing demonstrated the presence of the missense mutation-R342Q-consistent with the diagnosis of heterozygous Fabry disease. A renal biopsy specimen showed a total of 58 glomeruli, of which 8 (14%) were globally sclerotic and 7 (12%) revealed focal segmental sclerosis in association with Fabry disease. Fine foamy vacuolation was observed in the glomerular podocytes, tubular epithelium, and interstitial cells; this finding was suggestive of Fabry disease. Similar ultrastructural features, such as myelin figures as seen in Fabry disease, may be observed with exposure to chloroquine, aminoglycosides, laxatives, and silicon.
Currently, there are 2 forms of intravenous enzyme replacement therapy, agalsidase alfa and agalsidase beta.18 The treatment regimen and duration continue to be debated.
References:
References1. Shimazu K, Tomiyoshi Y, Aoki S, et al. Crescentic glomerulonephritis in a patient with heterozygous Fabry's disease. Nephron. 2002;92:456-458.
2. Rahman P, Gladman DD, Wither J, Silver MD. Coexistence of Fabry's disease and systemic lupus erythematosus. Clin Exp Rheumatol. 1998;16:475-478.
3. Rosenmann E, Kobrin I, Cohen T. Kidney involvement in systemic lupus erythematosus and Fabry's disease. Nephron. 1983;34:180-184.
4. Brady RO, Gal AE, Bradley RM, et al. Enzymatic defect in Fabry's disease: ceramidetrihexosidase deficiency. N Engl J Med. 1967;276:1163-1167.
5. Meroni M, Sessa A, Battini G, et al. Kidney involvement in Anderson-Fabry disease. Contrib Nephrol. 1997; 122:178-184.
6. Sheth KJ, Roth DA, Adams MB. Early renal failure in Fabry's disease. Am J Kidney Dis. 1983;2:651-654.
7. Sirvent AE, Enriquez R, Antolin A, et al. Fabry's disease presenting with oliganuric end-stage renal failure. Nephrol Dial Transplant. 1997;12:1503-1505.
8. Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med.2007;9:34-45.
9. Wilcox WR, Oliveira JP, Hopkin RJ, et al; Fabry Registry. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93:112-128.
10. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769-775.
11. Street NJ, Yi MS, Bailey LA, Hopkin RJ. Comparison of health-related quality of life between heterozygous women with Fabry disease, a healthy control population, and patients with other chronic disease. Genet Med. 2006;8:346-353.
12. Van Loo A, Vanholder R, Madsen K, et al. Novel frameshift mutation in a heterozygous woman with Fabry's disease and end-stage renal failure. Am J Nephrol. 1996;16:352-357.
13. Singh HK, Nickeleit V, Kriegsmann J, et al. Coexistence of Fabry's disease and necrotizing and crescentic glomerulonephritis. Clin Nephrol. 2001;55:73-79.
14. Clarke JT. Narrative review: Fabry disease. Ann Intern Med. 2007;146:425-433.
15. McAlarney T, Ogino M, Apostolski S, Latov N. Specificity and cross-reactivity of anti-galactocerebroside antibodies. Immunol Invest. 1995;24:595-606.
16. Kasai N, Pachner AR, Yu RK. Anti-glycolipid antibodies and their immune complexes in multiple sclerosis. J Neurol Sci. 1986;75:33-42.
17. Gupta S, Ries M, Kotsopoulos S, Schiffmann R. The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. Medicine (Baltimore). 2005;84:261-268.
18. Torra R. Renal manifestations in Fabry disease and therapeutic options. Kidney Int Suppl. 2008;111:S29-S32.














