Systemic Lupus Erythematosus With Features of Castleman Disease
SLE can manifest as lymphadenopathy. This man fulfilled at least 4 diagnostic criteria for SLE, including rash and blood results.

A 49-year-old African American man was referred to rheumatology with a positive antinuclear antibodies (ANA) test result, constitutional symptoms, and lymphadenopathy. He presented with unintentional weight loss, low-grade fever, night sweats, and malaise. The patient also complained of pain in several joints and a history of minimal sun exposure that caused a painful rash.
The physical examination revealed that the patient had a low-grade fever, with a temperature of 37.7ºC (99.9ºF). His weight was 205 lb, down from 235 lb 6 months earlier. Widespread lymphadenopathy was found in the cervical, supraclavicular, axillary, and inguinal regions. Findings from the remainder of the examination were unremarkable, with no synovitis or rash. Laboratory findings included positive results for ANA (1:640, speckled pattern), anti-Smith antibodies, and anti-ribonucleoprotein antibodies; hypocomplementemia; polyclonal hypergammopathy; leukopenia; anemia; and elevated levels of inflammatory markers.
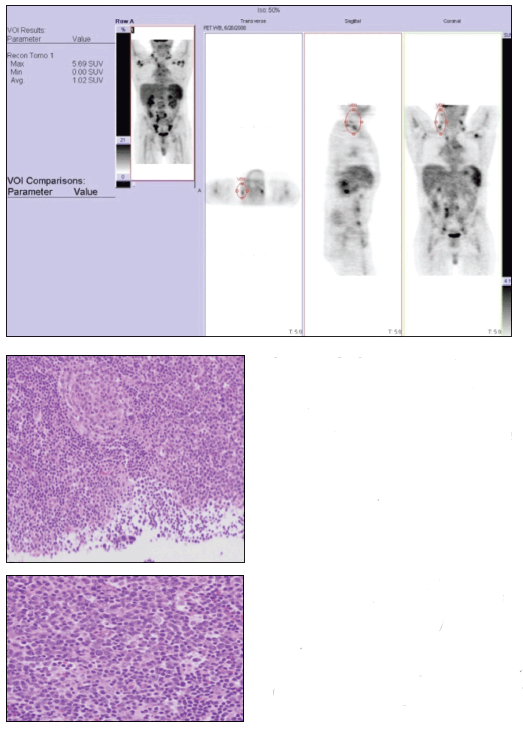
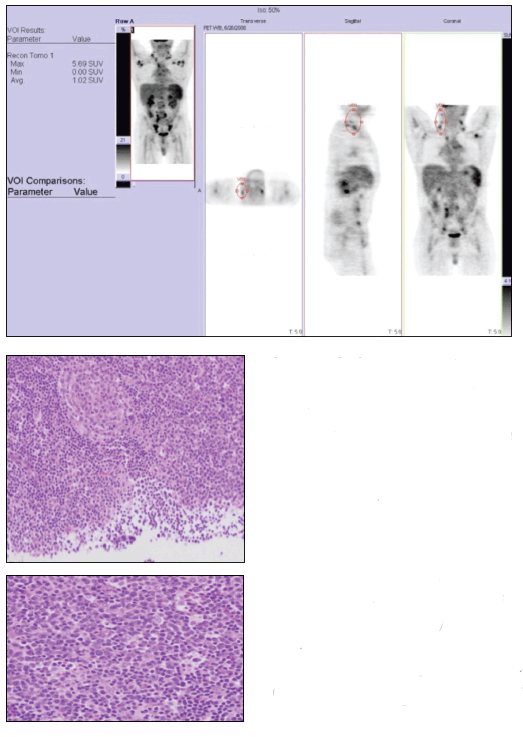
A whole-body positron emission tomography (PET) survey scan (top) demonstrated the diffuse nature of the patient’s lymphadenopathy. A right-sided supraclavicular lymph node excisional biopsy specimen revealed follicular hyperplasia and regressed follicles that were atretic with hyaline-vascular change, with sheets of plasma cells in the interfollicular zones (middle and bottom).
The diagnosis was systemic lupus erythematosus (SLE) manifesting as lymphadenopathy, with features of Castleman disease (CD).
The whole-body PET scan revealed diffuse hypermetabolic adenopathy extending from the neck through the chest and diffusely throughout the retroperitoneal and inguinal regions. The patient fulfilled at least 4 diagnostic criteria for SLE-photosensitive rash, positive test results for ANA and anti-Smith antibodies, and leukopenia. The diffuse lymphadenopathy, histologically marked by follicular hyperplasia and sheets of plasma cells, is consistent with CD, which was present in our patient with a new diagnosis of SLE.
Dr Benjamin Castleman1 first described CD in 1956 as a benign mediastinal lymph node hyperplasia resembling thymoma. A somewhat rare disorder, CD is characterized by a massive nonmalignant tumor of lymphoid tissues; the cause is unknown.2
CD has unicentric and multicentric forms. Most cases of unicentric CD are asymptomatic and present with isolated lymphadenopathy. Although unicentric disease typically is considered benign, both unicentric and multicentric CD have associations with non-Hodgkin lymphoma.3,4 The multicentric form of CD affects more than a single group of lymph nodes, often presents with systemic symptoms, and has been linked with HIV-positive patients and with human herpes virus 8 (HHV8) infection.

Within those forms, CD has 3 major histological variants: hyaline vascular, plasma cell, and mixed. The hyaline vascular type is the most common, accounting for 80% of cases, and typically is seen in the localized form; the plasma cell type is less common and usually is more aggressive.
The systemic manifestations of fever, anemia, elevated erythrocyte sedimentation rate, and hypergammaglobulinemia occur frequently in patients who have the plasma cell type of CD. These systemic symptoms are particularly relevant to this discussion because some rheumatologic disorders-rheumatoid arthritis, various connective-tissue diseases, and SLE-may cause hyperplastic reactive lymph nodes with increased plasma cells.2
The differential diagnosis for diffuse lymphadenopathy is extensive, including but not limited to infection, malignancy, and autoimmune or connective-tissue disease. Excluding reversible causes of lymphadenopathy is imperative.
The workup for our patient yielded negative results for infection and malignancy. Of note, he had negative HIV and HHV8 test results; bone marrow biopsy, colonoscopy, and esophagogastroduodenoscopy results were normal.
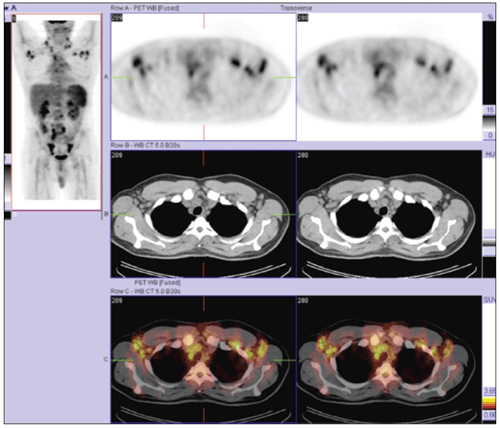
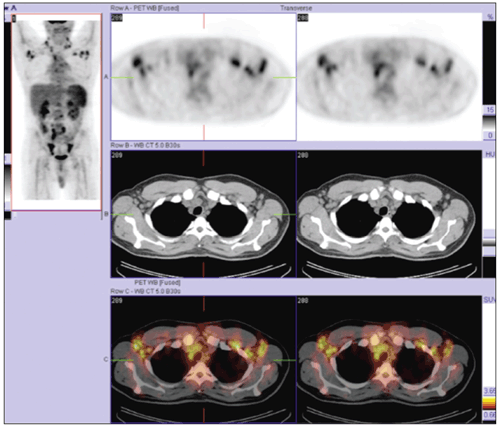
If the initial workup does not provide a diagnosis in cases of diffuse lymphadenopathy, tissue architecture analysis often is required.2,5 Our patient’s PET/CT fusion images (above) revealed lymphadenopathy and made it easily accessible to surgeons and interventional radiologists. Our excisional biopsy eventually directed the diagnosis and treatment.
SLE has 2 major histological patterns, coagulative necrosis with hematoxylin bodies and reactive follicular hyperplasia. Coagulative necrosis is very specific for SLE but has poor sensitivity. Reactive follicular hyperplasia has multiple subtypes, one of which is CD.
In a retrospective review of 33 cases of SLE with lymphadenopathy at any time during the disease course, only 5 cases showed histological features consistent with CD.6 The presence of lymphadenopathy at SLE diagnosis is even more rare; there are few documented cases.7,8 The cumulative incidence of lymphadenopathy at some point in SLE disease progression is high, up to 60%.2
In a 2010 case report and literature review, Xia and colleagues2 stated that the specific histological finding of lymph node necrosis is actually quite rare in SLE lymphadenopathy, follicular hyperplasia usually is considered nonspecific, and features consistent with CD often are overlooked. We agree with their conclusion that considering the possibility of CD in patients with SLE lymphadenopathy is important.
Treatment of patients with CD varies with the specific form, histological variant, and comorbidities. The unicentric form often is managed successfully with resection. The multicentric form may be managed with a combination of surgery, high-dose corticosteroids, and systemic chemotherapy.
Our patient was treated with high-dose oral prednisone, with a long taper. Hydroxychloroquine was started early and remained a staple of his therapy. His laboratory test results normalized, and the constitutional symptoms, rash, and arthralgia resolved. At 3 years out, he remains symptom-free.
References:
REFERENCES
1. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956;9:822-830.
2. Xia JY, Chen XY, Xu F, et al. A case report of systemic lupus erythematosus combined with Castleman’s disease and literature review. Rheumatol Int. 2010 Mar 31; [Epub ahead of print]. Accessed June 23, 2011. doi:10.1007/s00296-010-1451-0.
3. Herrada J, Cabanillas F, Rice L, et al. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med. 1998;128:657-662.
4. Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29:670-683.
5. Kim KJ, Cho CS, Choi JJ. Pararenal retroperitoneal Castleman’s disease mimicking systemic lupus erythematosus. Int J Rheum Dis. 2010;13:e20-e25.
6. Kojima M, Motoori T, Asano S, Nakamura S. Histological diversity of reactive and atypical proliferative lymph node lesions in systemic lupus erythematosus patients. Pathol Res Pract. 2007;203:423-431.
7. Suwannaroj S, Elkins SL, McMurray RW. Systemic lupus erythematosus and Castleman’s disease. J Rheumatol. 1999;26:1400-1403.
8. Van de Voorde K, De Raeve H, De Block CE, et al. Atypical systemic lupus erythematosus or Castleman’s disease. Acta Clin Belg. 2004;59:161-164.














