Current concepts in corticosteroid-induced bone disease
The most common secondary form of osteoporosis is corticosteroid-induced disease. Cases of endogenous corticosteroid excess are few compared with the large number produced by exogenous use of the drug for management of many diverse diseases.
Corticosteroid-induced osteoporosis is one of the most common secondary causes of osteoporosis. Published treatment guidelines attest to worldwide concern about the problem.1-3 The diagnoses that most often are associated with corticosteroid use are chronic obstructive pulmonary disease, asthma, and rheumatoid arthritis.4 Although great advances have been made in the past decade or so in understanding the epidemiology, pathology, diagnosis, and management of the problem, medical practice still misses a large number of patients who need treatment.
A recurring theme is seen in large epidemiological studies of managed care databases. Fewer than 50% of patients who are receiving therapy with corticosteroids have adequate instruction in preventive measures for osteoporosis, and even fewer actually receive some form of useful therapy. Measurements of bone mass are made in about 10% of patients, and only 38% of patients receive any treatment.4
There is reason for optimism. In managed care databases, bone measurements increased about 3-fold in 2001-2003 compared with 1995-1998, and the use of prescription antiresorptive medications increased about 2-fold.5 Although busy physicians in family or general practice were less likely than other physicians to request measurements of bone mass or prescribe therapy, and counseling of patients was limited,6 concerted efforts to address these problems are effective. Educational programs that focused on the activities of general practitioners and pharmacists in Australia showed substantial improvement in preventive therapy.7
The means are available to provide appropriate care for patients with corticosteroid-induced osteoporosis. The real issue is a lack of implementation. In this article, I touch on paradoxes in diagnosis of this disorder by bone mass measurements, review some new concepts in the pathophysiology, and discuss current and future therapies.
Diagnosis
Making a diagnosis of osteoporosis in patients who are using corticosteroids is not as clear-cut as might be expected. Clinicians recognize that bone densitometry is singularly effective in estimating fracture risk when used for the patient population for which it was developed, older postmenopausal Caucasian women. The “magical” T-score of −2.5 standard deviations or lower is ingrained in the minds of physicians as a surrogate marker for osteoporosis and risk of fragility fractures. When this

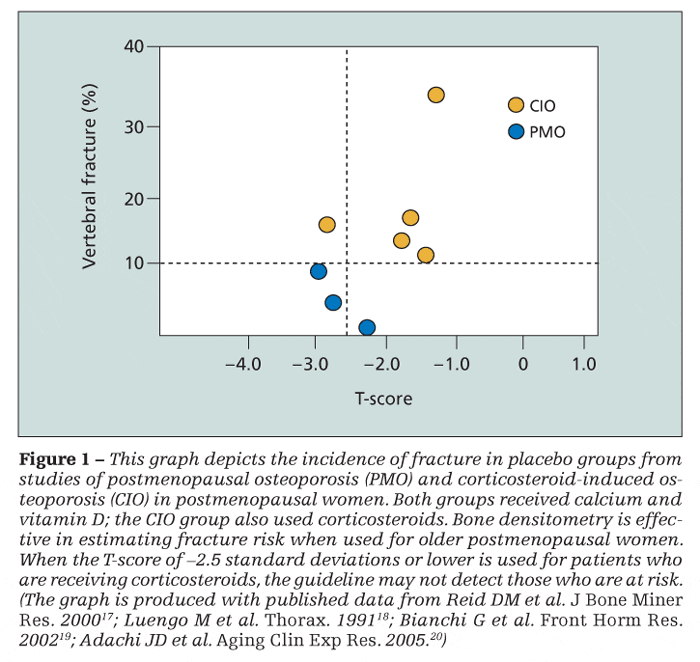
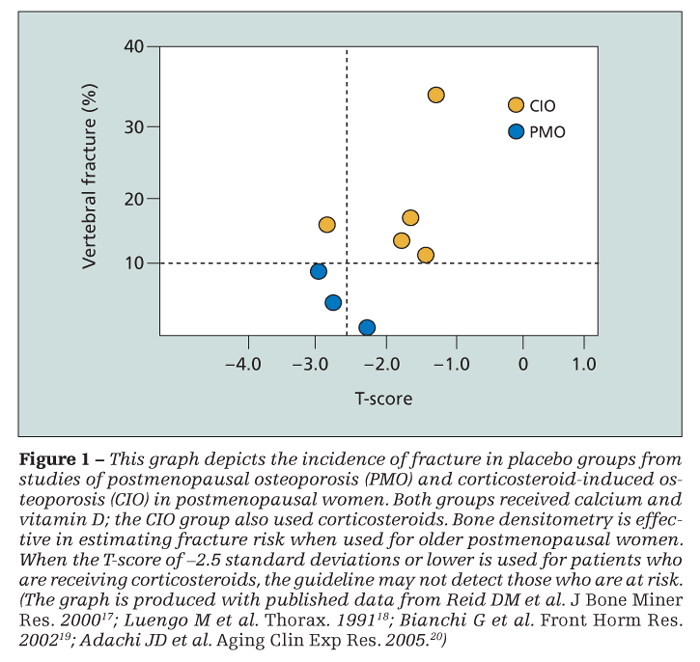
T-score is used for patients who are receiving corticosteroids, however, the guideline may not detect those who are at risk (Figure 1). The prevalence of vertebral fracture is greater in patients who are using oral corticosteroids, and they tend to fracture at a greater bone mass.8
Some authors argue that these patients fracture at the same density or T-score regardless of corticosteroid use.9 However, clinical experience shows that these opinions rest on population data, which may not be useful in daily practice; such notions provide little confidence that a patient who has “good density” will not fracture. Although guidelines focus attention on long-term use of oral corticosteroids at a dosage of 7.5 mg/d of prednisone or the equivalent for 3 months or more, fracture risk is increased at any dosage, however small.10
Inhaled corticosteroids are another risk factor for bone loss. Some potent forms (eg, fluticasone propionate) may have systemic effects.
Why the conundrum in clinical bone density measurements and fracture risk? Corticosteroids are causing changes in bone strength that are not related to density alone. The quality of the structure is compromised; problems may be detected with sophisticated engineering techniques.11
Dual-energy x-ray absorptiometry (DXA) is the gold standard for noninvasive measurement of bone mass in various anatomical sites. DXA involves low radiation exposure, and the machines are somewhat inexpensive. The World Health Organization criteria for T-scores and the guidelines for making a diagnosis of osteoporosis derive from this technology.
Quantitative CT (QCT) arose as a research tool for making more precise measurements of true volumetric bone density but in the vertebrae only. QCT involves high radiation exposure, and the machines (CT scanners) are expensive. Measurements of mass with QCT are seen occasionally in clinical practice, but they have not been standardized in practice as true DXA analyses have. This deficiency leads to greater measurement error from one scan to the next.
Pathophysiology
Corticosteroids have a variety of effects on the organ systems within the body; therefore, they cause osteoporosis through a variety of mechanisms (Table 1).12 Some of the mechanisms may



not adequately explain what is known about the disease process right now, but they are still discussed in the literature.
The least understood cause of osteoporosis in patients who use corticosteroids is the underlying disease for which the drugs were originally prescribed. In inflammatory bowel disease, for example, the inflammation itself may have a negative impact on bone metabolism apart from the use of corticosteroids.
Corticosteroids add to the negative effects of any disease. They impair the action of vitamin D on intestinal calcium absorption and increase renal loss of calcium. Both effects lower the ambient serum calcium level and trigger a secondary hyperparathyroid state that might increase bone resorption. However, this is a subject of controversy and data are lacking.
Corticosteroids impair pituitary secretion of gonadotrophins, in turn decreasing gonadal hormones in men and women. This “chemical hypogonadism” enhances osteoclastic activity and subsequent skeletal destruction. Recent experimental data show that these drugs inhibit vascular endothelial growth factor, a protein involved in ossification.13
Apart from all of this physiological change, there are direct skeletal effects on bone cells. Aging (apoptosis) of osteoclastic cells changes with corticosteroid use. Corticosteroids increase expression of RANKL (receptor activated nuclear factor-κB ligand) and decrease expression of osteoprotegerin. Both actions promote osteoclastogenesis and osteoclast lifespan. The cells age more slowly and live longer. Conversely, osteoblasts age at a faster rate and die sooner. The osteocyte, which maintains normal adult bone, also may be affected. Its early death may cause bone destruction and may be an integral component of osteonecrosis.14
Histological changes in corticosteroid-treated bone are different from those in postmenopausal osteoporosis. Trabecular thinning and perforations are common in primary disease; with corticosteroid use, perforations are uncommon but thinning still occurs.
Treatment
Published guidelines for treatment are similar across all subspecialties. Patients should start therapy for prevention or management of osteoporosis if use of 7.5 mg/d of prednisone or its

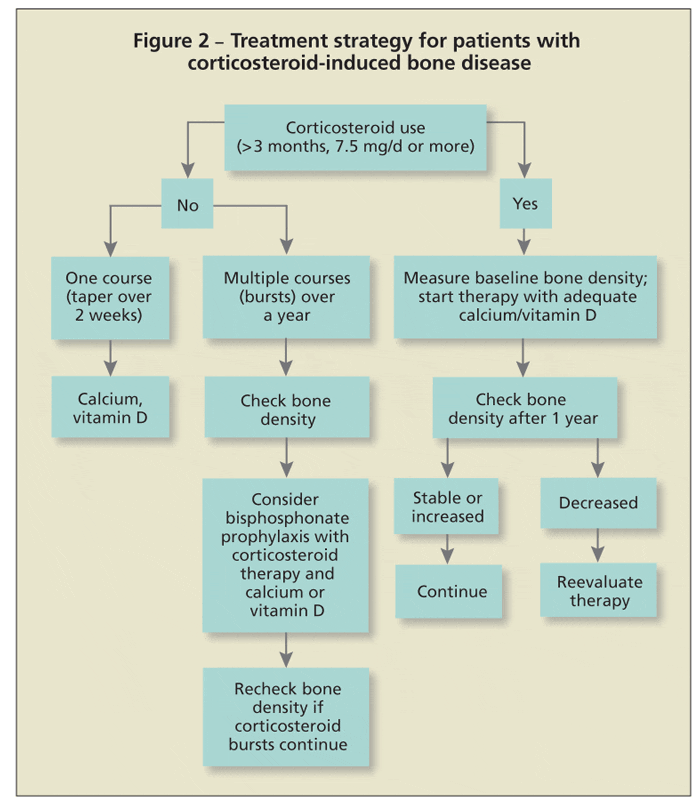
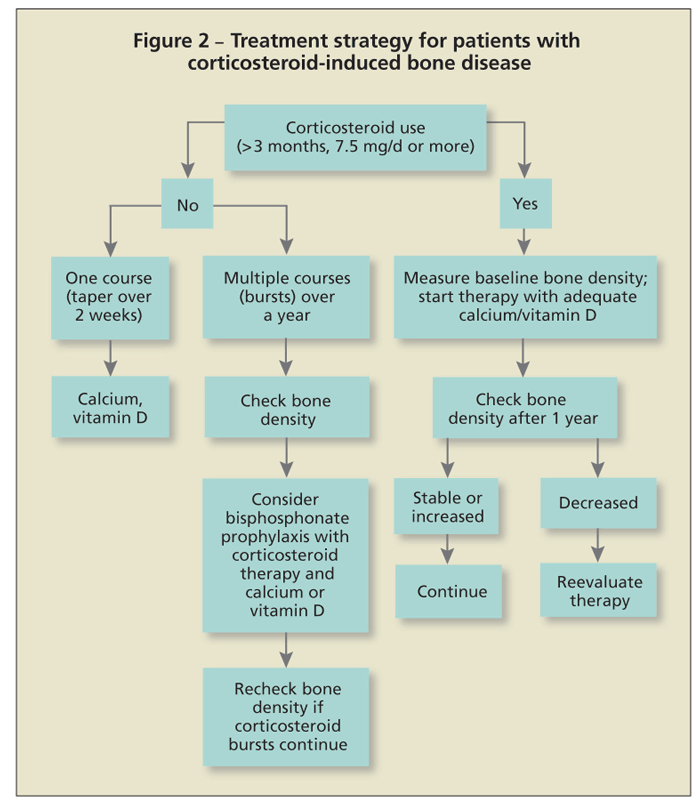
equivalent for 3 months or longer is anticipated (Figure 2). A baseline bone density measurement will indicate the status of bone mass. Negative T-scores or Z-scores of 2 or higher are compelling reasons for physicians to institute treatment.
The problem, however, is what to do in cases of normal scores. Because corticosteroids can cause fractures even with normal bone mass, therapeutic intervention may be necessary. Most bone loss occurs quickly in the first 6 to 12 months after the patient starts taking corticosteroids. Bone metabolism (turnover) is high, as evidenced with measurements of bone turnover markers. If used rarely, short courses of corticosteroid therapy (eg, 2-week dose packs) should not be a major concern. Repeated use of such courses or, more commonly, repeated use of injectable corticosteroids warrants consideration of prophylactic skeletal therapy.
A variety of treatment options have become available for osteoporosis prevention or management or both (Table 2), but only bisphosphonates and the anabolic drug teriparatide are FDA-approved for this use. Intravenous zoledronic acid is used yearly for this purpose. The injectable drug

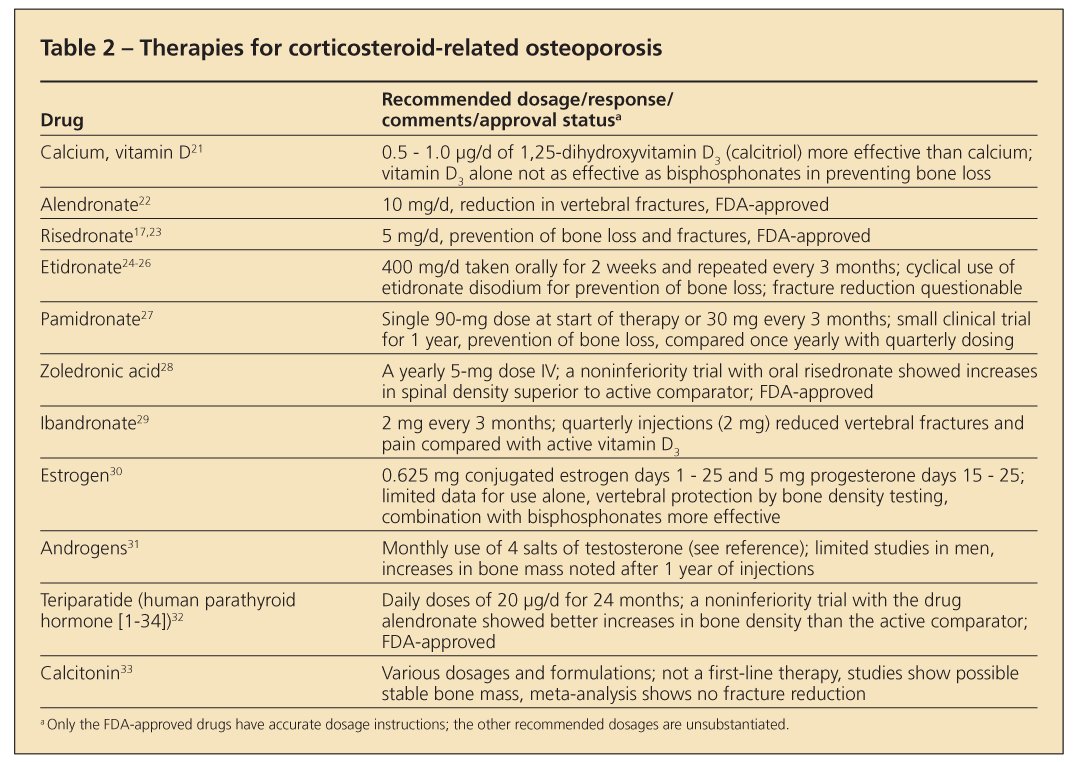
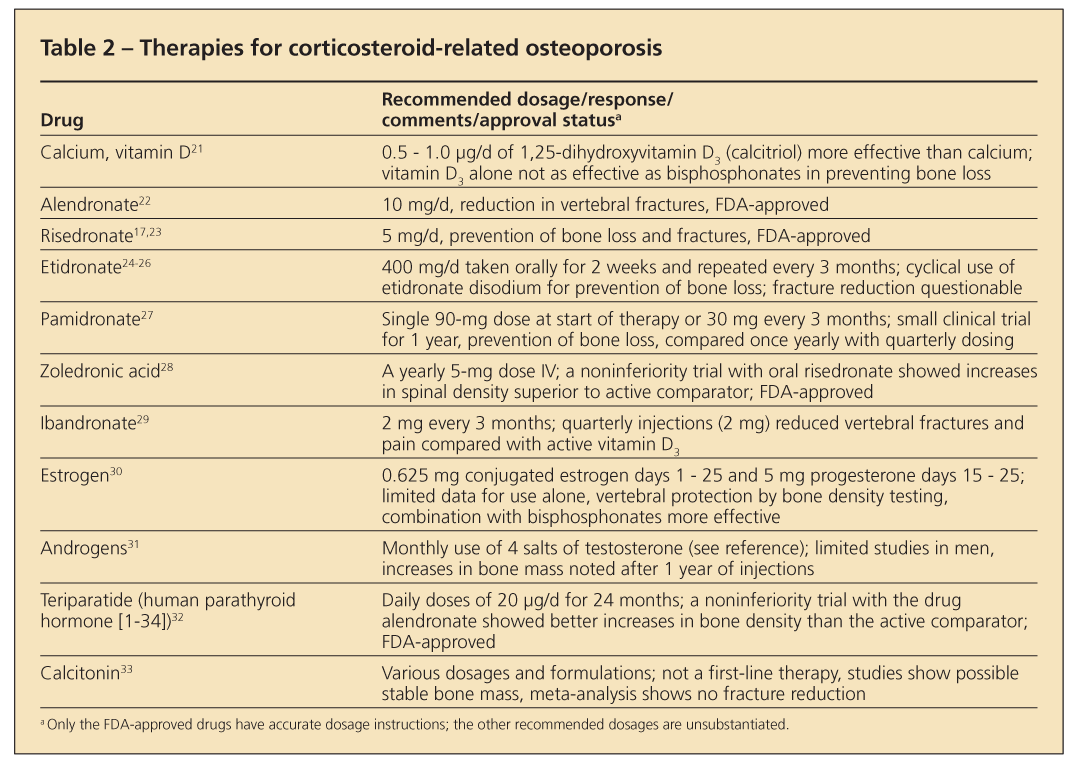
teriparatide is used daily for 24 months by patients.15,16 Early therapies were directed at physiological sites of adverse corticosteroid effects. Because of limited statistical power, most investigations monitored changes in bone density only and did not address fracture reduction, the present-day standard of evaluating FDA-approved treatments.
Use of calcium and large doses of vitamin D was the hallmark of early therapeutic efforts to compensate for poor absorption of calcium and secondary hyperparathyroidism. However, success was limited. Corticosteroids have a direct effect on intestinal calcium absorption, which may not correct with intake of additional calcium and vitamin D.
Hydrochlorothiazide was used to suppress hypercalciuria and promote a more positive calcium balance and maintain bone mass. Exogenous estrogens and androgens rounded out treatment regimens and helped correct the hypogonadal state. However, they also had limited success. Looking at this therapy from a skeletal pathology point of view, the approach was meant to slow bone loss (antiresorption) or build new bone (anabolism); efficacy was limited.
The antiresorptive drugs calcitonin and etidronate disodium-the first drug in the bisphosphonate class-provided approaches to controlling osteoclastic activity. Both drugs prevent some bone loss.
With the arrival of the FDA-approved bisphosphonates alendronate and risedronate, significant effects on fracture prevention and reduction became a reality. These agents show similar clinical effects. Other bisphosphonates (eg, pamidronate, zoledronic acid, and ibandronate) show potential for effective treatment. The literature references their use, but formal FDA approval is available only for zoledronic acid.
The drug teriparatide (human parathyroid hormone [1-34]) is the first in a new class of drugs called anabolic agents that has received FDA approval for osteoporosis use. Teriparatide increases skeletal mass after a year of use in patients who are using prednisone, but as yet there are no fracture data.



Further evaluations of this drug class probably are forthcoming. Sodium fluoride met the need for an agent to enhance bone formation, but concern about its skeletal toxicity limited its use.
Studies of FDA-approved bisphosphonates show a reduction in fractures in patients who are treated with corticosteroids. In addition, the drugs can stabilize bone and prevent density loss in patients who are starting therapy.
Conclusions
The current challenge that corticosteroid-induced osteoporosis presents to patients and clinicians is not a lack of treatment options. Ample numbers of bisphosphonates hold the unique potential for providing efficacious treatment, but the new anabolic class of drugs offers a new approach to treating the problem, which may prove over time to be more effective. A lack of implementing what is available to diagnose and manage the problem is the real issue. The reason for this inertia is unclear.
Merely developing more therapeutic options does little to improve understanding of why the available drugs are rarely used. Future clinical studies must address this issue, and improved education should be a component of future efforts. Perhaps developing more convenient types of treatment would help.
Until the use of corticosteroids is recognized as a problem that should be managed, however, advances in diagnosis, understanding of the pathology, and therapy will be a moot issue. The means to care for patients appropriately is here. The initiative for success is in the hands of clinicians.
References:
References
1. Geusens PP, de Nijs RN, Lems WF, et al. Prevention of glucocorticoid osteoporosis: a consensus document of the Dutch Society for Rheumatology. Ann Rheum Dis. 2004;63:324-335.
2. Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of The Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab. 2005;23:105-109.
3. Compston J. US and UK guidelines for glucocorticoid-induced osteoporosis: similarities and differences. Curr Rheumatol Rep. 2004;6:66-69.
4. Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int. 2005;16:2168-2174.
5. Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum. 2005;52:2485-2494.
6. Blalock SJ, Norton LL, Patel RA, Dooley MA. Patient knowledge, beliefs, and behavior concerning the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Rheum. 2005;53:732-739.
7. Naunton M, Peterson GM, Jones G, et al. Multifaceted educational program increases prescribing of preventive medication for corticosteroid induced osteoporosis. J Rheumatol. 2004;31:550-556.
8. Peel NF, Moore DJ, Barrington NA, et al. Risk of vertebral fracture and relationship to bone mineral density in steroid treated rheumatoid arthritis. Ann Rheum Dis. 1995;54:801-806.
9. Evans SF, Davie MW. Vertebral fractures and bone mineral density in idiopathic, secondary and corticosteroid associated osteoporosis in men. Ann Rheum Dis. 2000;59:269-275.
10. Van Staa TP, Leufkens HG, Abenhaim L, et al. Use of oral corticosteroids and risk of fractures. J Bone Miner Res. 2000;15:993-1000.
11. Lian KC, Lang TF, Keyak JH, et al. Differences in hip quantitative computed tomography (QCT) measurements of bone mineral density and bone strength between glucocorticoid-treated and glucocorticoid-naive postmenopausal women. Osteoporos Int. 2005;16: 642-650.
12. Dalle Carbonare L, Bertoldo F, Valenti MT, et al. Histomorphometric analysis of glucocorticoid-induced osteoporosis. Micron. 2005;36:645-652.
13. Pufe T, Scholz-Ahrens KE, Franke AT, et al. The role of vascular endothelial growth factor in glucocorticoid-induced bone loss: evaluation in a minipig model. Bone. 2003;33:869-876.
14. Zalavras C, Shah S, Birnbaum MJ, Frenkel B. Role of apoptosis in glucocorticoid-induced osteoporosis and osteonecrosis. Crit Rev Eucharyot Gene Expr. 2003;13:221-235.
15. Saag KG, Zanchetta JR, Devogelaer JP, et al. Effects of teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: thirty-six-month results of a randomized, double-blind, controlled trial. Arthritis Rheum. 2009;60:3346-3355.
16. Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet. 2009;373:1253-1263.
17. Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-induced Osteoporosis Treatment Study. J Bone Miner Res. 2000;15:1006-1013.
18. Luengo M, Picado C, Del Rio L, et al. Vertebral fractures in steroid dependent asthma and involutional osteoporosis: a comparative study. Thorax. 1991;46:803-806.
19. Bianchi G, Girasole G, Sinigaglia L. DXA in the diagnosis of glucocorticoid-induced osteoporosis. Front Horm Res. 2002;30:13-19.
20. Adachi JD, Rizzoli R, Boonen S, et al. Vertebral fracture risk reduction with risedronate in post-meno-pausal women with osteoporosis: a meta-analysis of individual patient data. Aging Clin Exp Res. 2005;17:150-156.
21. de Nijs RN, Jacobs JW, Algra A, et al. Prevention and treatment of glucocorticoid-induced osteoporosis with active vitamin D3 analogues: a review with meta-analysis of randomized controlled trials including organ transplantation studies. Osteoporos Int. 2004;15:589-602.
22. Adachi JD, Saag KG, Delmas PD, et al. Two-year effects of alendronate on bone mineral density and vertebral fracture in patients receiving glucocorticoids: a randomized, double-blind, placebo-controlled extension trial. Arthritis Rheum. 2001;44:202-211.
23. Eastell R, Devogelaer JP, Peel NF, et al. Prevention of bone loss with risedronate in glucocorticoid-treated rheumatoid arthritis patients. Osteoporos Int. 2000;11:331-337.
24. Adachi JD, Bensen WG, Brown J, et al. Intermittent etidronate therapy to prevent corticosteroid-induced osteoporosis. N Engl J Med. 1997;337:382-387.
25. Roux C, Oreinte P, Laan R, et al. Randomized trial of effect of cyclical etidronate in the prevention of corticosteroid-induced bone loss. Ciblos Study Group. J Clin Endocrinol Metab. 1998;83:1128-1133.
26. Campbell IA, Douglas JG, Francis RM, et al; Research Committee of the British Thoracic Society. Five year study of etidronate and/or calcium as prevention and treatment for osteoporosis and fractures in patients with asthma receiving long-term oral and/or inhaled glucocorticoids. Thorax. 2004;59:761-768.
27. Boutsen Y, Jamart J, Esselinckx W, Devogelaer JP. Primary prevention of glucocorticoid-induced osteoporosis with intravenous pamidronate and calcium: a prospective controlled 1-year study comparing a single infusion, an infusion given once every 3 months, and calcium alone. J Bone Miner Res. 2001;16:104-112.
28. Souza SC, Borges CT, Jorgetti V, Pereira RM. The effect of intravenous zoledronic acid on glucocorticoid-induced multiple vertebral fractures in juvenile systemic lupus erythematosus. Rev Hosp Clin Fac Med Sao Paulo. 2004;59:302-305.
29. Ringe JD, Dorst A, Faber H, et al. Intermittent intravenous ibandronate injections reduce vertebral fracture risk in corticosteroid-induced osteoporosis: results from a long-term comparative study. Osteoporos Int. 2003;14:801-807.
30. Lukert BP, Johnson BE, Robinson RG. Estrogen and progesterone replacement therapy reduces glucocorticoid-induced bone loss. J Bone Miner Res. 1992;7:1063-1069.
31. Reid IR, Wattie DJ, Evans MC, Stapleton JP. Testosterone therapy in glucocorticoid-treated men. Arch Intern Med. 1996;156:1173-1177.
32. Lane NE, Sanchez S, Modin GW, et al. Bone mass continues to increase at the hip after parathyroid hormone treatment is discontinued in glucocorticoid-induced osteoporosis: results of a randomized controlled clinical trial. J Bone Miner Res. 2000;15:944-951.
33. Cranney A, Welch V, Adachi JD, et al. Calcitonin for the treatment and prevention of corticosteroid-induced osteoporosis. Cochrane Database Syst Rev. 2000;2:CD001983.
Recommended reading
The author suggests the following source for readers seeking more information:
•Giustina A, Angeli A, Canalis E, Manelli F, eds. Glucocorticoid-Induced Osteoporosis. Basel, NY: Karger AG; 2002;30:1-180. This compendium of journal articles covering the pathology, diagnosis, and treatment is a useful reference for clinicians.














