Decision Making in Systemic Sclerosis: A Guide for Primary Care
The name and identifying features derive from what happens to the skin: it becomes thickened and swollen from deposition of excessive amounts of collagen.
The name and identifying features of systemic sclerosis (SSc), a systemic illness, derive from what happens to the skin: it becomes thickened and swollen (fibrosed, or sclerotic) from deposition of excessive amounts of collagen. In some cases, as in diffuse SSc, skin involvement is the major cause of morbidity. However, the degree of morbidity and mortality in SSc often is related more to coexisting internal involvements-heart, lung, kidney, GI, and peripheral vascular.
Assessing skin thickening and determining how long the patient has had SSc are major points in disease management. Because major organ involvements usually begin and progress most rapidly early in the disease, they should be detected early so that treatment can be instituted as soon as possible.
Many patients with SSc present at their initial visit terrified by misinformation acquired through friends, relatives, and the Internet. To optimize patient care, the physician should spend extra time discussing the disease with patients and educating them so that they understand it and can become active participants in their evaluation and treatment.
SSc is a somewhat uncommon disease. It has an annual incidence of 20 cases per million persons in the United States; the prevalence is about 240 cases per million persons.1 Peak onset is at about age 50 years in the United States. SSc affects 3 to 4 times as many women as men. The rate of mortality is thought to be higher in men and in African Americans than in others.1

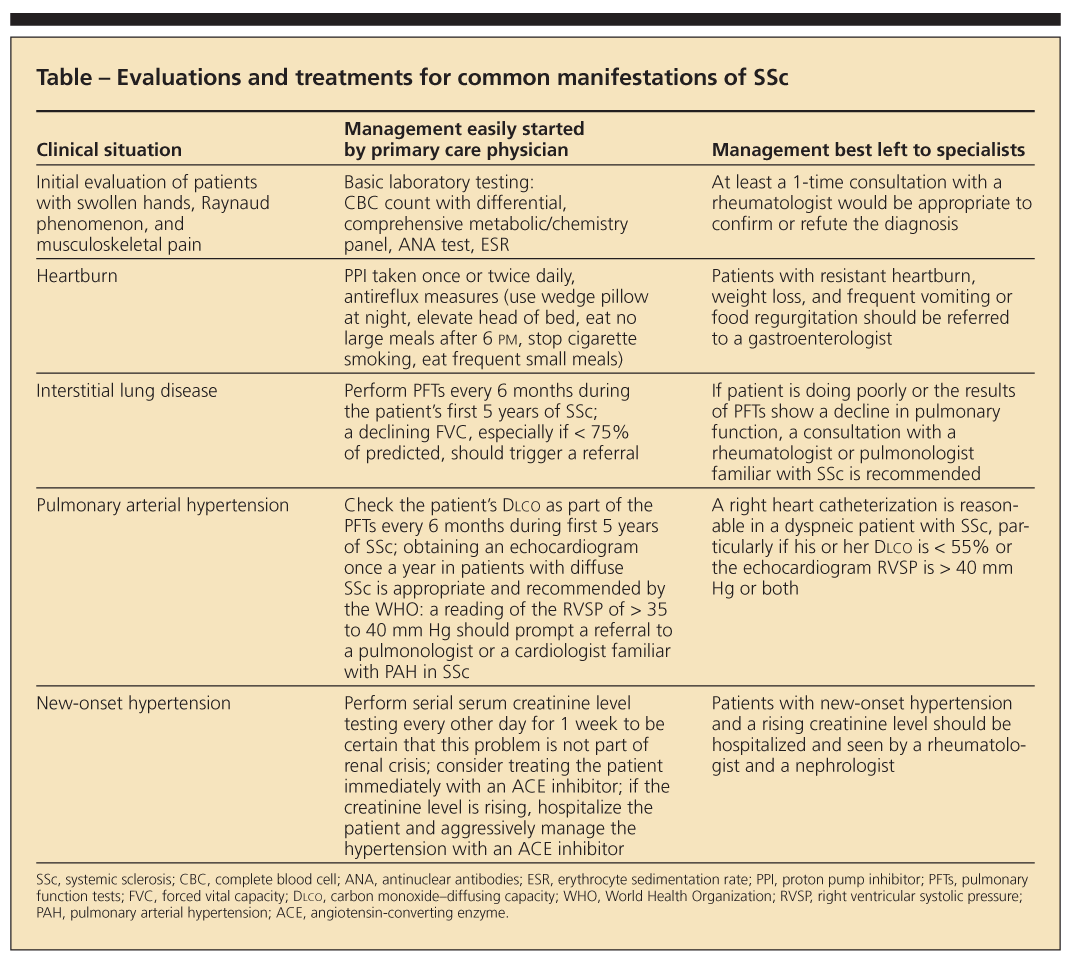
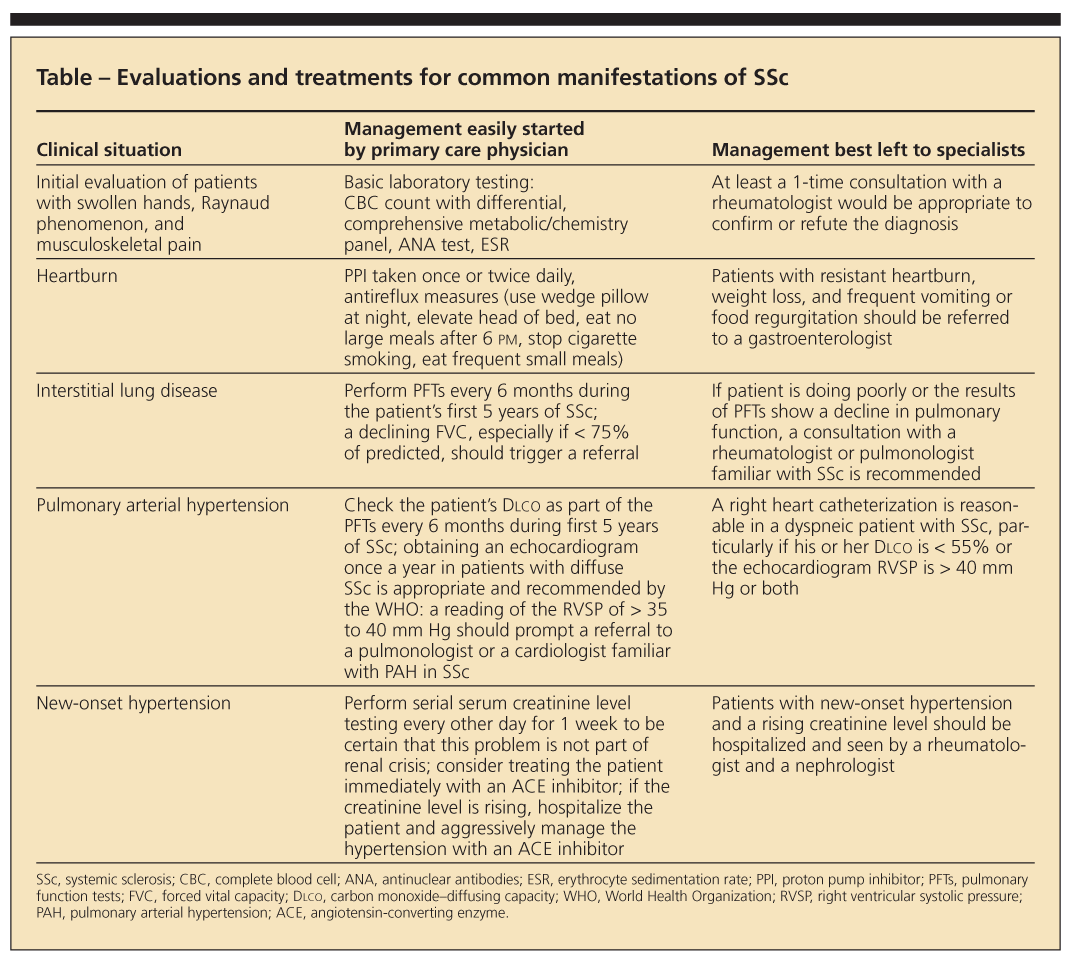
Because SSc is uncommon, primary care physicians do not see patients with this disease very often. However, they may easily start some evaluations and treatments for common manifestations of SSc and refer other patients to specialists (Table). In this article, we attempt to attune primary care physicians to the initial diagnosis and to the most pressing questions they and their patients will confront so that together they can make the most appropriate decisions.
THE QUESTIONS
1. What in the patient's history and my physical examination should make me consider a diagnosis of SSc?
Most patients with SSc indicate that their initial symptoms were one of the following:
•The new onset of intermittent color changes in the fingers, usually in response to cold exposure or emotion (Raynaud phenomenon), most frequently to blue and white (Figure 1).
•Finger swelling.
•The onset of musculoskeletal pain, which can be diffuse, in muscles, or associated with multiple joints (polyarthralgia or polyarthritis or both) and often is associated with 30 or more minutes of morning stiffness.
Patients may have these complaints early on. At this juncture, however, they may not have other characteristic signs and symptoms that would make the diagnosis of SSc clear-cut.
Figure 1 – The cyanotic phase of Raynaud phenomenon is evident in color changes in several fingertips in a patient with systemic sclerosis.
Swelling, tightness, and thickness of the skin are the hallmarks of SSc. In some patients, these symptoms may be limited to the fingers; in others, they may involve the distal extremities and face (in either scenario, called "limited cutaneous scleroderma"). In about 40% of patients, the skin of the proximal extremities and the torso may become tight and thick (this is now called "diffuse cutaneous scleroderma").2
Once the physician is considering SSc, he or she should focus on the following features:
•Sclerodactyly (tight or thick skin of the fingers or both, particularly over the middle phalanges) occurs in 95% of patients with SSc (Figure 2).
•Raynaud phenomenon occurs in about 90% of patients.
•The antinuclear antibodies (ANA) test result is positive in 95% of patients with SSc.3

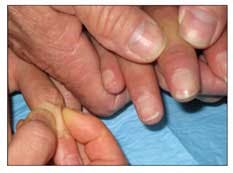
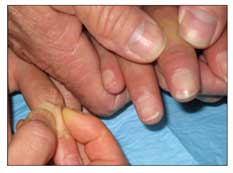
Figure 2 – Tight or thick skin of the fingers is a symptom of systemic sclerosis (SSc). The normal skin fold of a healthy person can be "pinched" over the middle phalanx (left), but in a patient with SSc (right), no skin fold can be pinched over the middle phalanx.
A patient who has none of these features rarely has SSc. If the patient's skin is thick elsewhere (eg, the extremities and torso) but not in the fingers, think of other disorders that can make the skin thick or tight (eg, eosinophilic fasciitis, nephrogenic systemic fibrosis, scleredema, and scleromyxedema). At this point, refer the patient to a knowledgeable dermatologist or rheumatologist.
2. Do the extent and degree of skin thickening provide the physician with information about prognosis?
Patients with SSc who are destined to have diffuse cutaneous thickening usually do so within 1 to 4 years after disease onset. Therefore, if a patient with limited SSc does not have an explosion of skin thickening into diffuse SSc within the first 3 to 4 years, it is unlikely that he will ever have diffuse SSc.
This distinction is important because patients with diffuse SSc are at risk for heart, lung, and kidney involvement-usually within the first 5 years of their SSc-and the mortality rate is generally higher for these patients. Conversely, patients with limited SSc are at low risk for heart or kidney involvement, although they and patients who have diffuse SSc are at nearly the same risk for interstitial lung involvement.2
3. Do rheumatologic disease blood tests help in making the diagnosis?
Because the ANA test result is positive in many patients who have other rheumatologic diseases, the test is not specific for SSc. Several autoantibody tests have more specificity for SSc, however, and are readily available in commercial laboratories.
Anticentromere antibodies occur in 40% to 50% of patients with limited SSc and 5% to 10% of those with diffuse SSc; significant interstitial lung disease (ILD) will develop in a very low percentage of patients who test positive.3,4 Conversely, antibodies against topoisomerase (Scl-70) occur in 30% to 35% of patients with diffuse SSc and 10% to 20% of patients with limited SSc; however, these patients are at risk for significant ILD. Anti-RNA polymerase III (RNA Pol III), a nucleolar antibody, occurs primarily in patients with diffuse SSc; in that population, it is associated with a 25% to 30% risk of renal crisis.
Many patients with SSc are not positive for anticentromere, Scl-70, or RNA Pol III antibodies, even though 95% of patients are positive for ANAs. Therefore, the diagnosis of SSc usually is made on the basis of physical findings on clinical examination.
4. What kinds of lung involvement are seen in SSc?
Two kinds of pulmonary disorders are typical in patients with SSc: ILD and pulmonary vascular disease, particularly pulmonary arterial hypertension (PAH). Together, these are the leading causes of death in patients with SSc.
As a result of interstitial fibrosis, about 40% of patients with SSc (both limited and diffuse) have a decline in their forced vital capacity (FVC) to less than 75% of predicted; FVC declines below 50% of predicted in only 10% to 15% of patients.5 The time during which the greatest loss of FVC occurs is within the first 4 to 6 years of SSc; in some patients, the loss happens quickly (20% or more of FVC may be lost within a 6-month period).5
Because patients may be not be symptomatic during this process (eg, no shortness of breath), physicians are strongly encouraged to perform pulmonary function tests (PFTs) (to include carbon monoxide–diffusing capacity [Dlco] and spirometry) every 6 months during the first 5 years of SSc. Testing proactively allows the physician to pick up any decline in FVC at the earliest possible time so that patients can be treated before their vital capacity drops to moderate (less than 75% of predicted) or severe (less than 50% of predicted) levels.

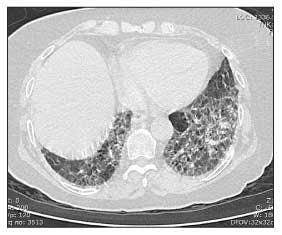
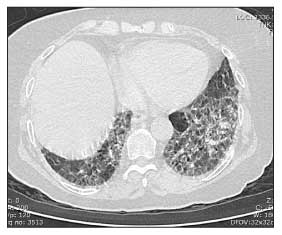
Figure 3 – In a patient with systemic sclerosis, a high-resolution CT scan of the chest may show fibrotic stranding in the posterior basilar segments bilaterally.
A high-resolution CT (HRCT) scan of the chest should be obtained if the patient's FVC drops below 80% of predicted or the patient reports dyspnea (Figure 3). The presence of fibrosis, with or without ground-glass opacification, on an HRCT scan confirms that the reduced FVC probably is caused by ILD rather than weak respiratory muscles or a tight chest wall resulting from fibrotic skin.
When analyzing PFTs, physicians should note the following issues:
The test should be performed on the same machine each time because of a high rate of variability (sometimes more than 10% of predicted) among machines.
The physician should assess the rate of decline rather than just look at absolute numbers (a patient whose FVC drops from 100% to 85% of predicted has a significant decline but, per the report, still has "normal" lung function).
Several treatments have been shown to be efficacious in managing the ILD of SSc (eg, cyclophosphamide) or show promise (eg, mycophenolate mofetil). Therefore, patients who have declining FVC or report dyspnea or persistent cough should be referred for evaluation and treatment to a rheumatologist or pulmonologist who has experience in managing ILD in SSc.6
5. What signs and symptoms should raise suspicion of PAH in a patient with SSc?
PAH is the second major type of pulmonary involvement in SSc.7 It tends to be associated with moderate or severe ILD that occurs early in SSc (both limited and diffuse subtypes), or it may occur late in limited SSc in isolation, without significant ILD.
PAH tends to make itself felt in fairly rapid fashion: patients often report that they have noticed progressive worsening in their shortness of breath over the previous few months. PFTs often show a diffusing capacity less than 50% to 55% of predicted, irrespective of the level of FVC.
Echocardiography may show an elevated right ventricular systolic pressure (RVSP), although it may overestimate or underestimate RVSP by at least 10 mm Hg in 50% of cases.8 RVSP readings between 35 and 45 mm Hg often are not accurate (when compared with right heart catheterization, the gold standard); levels higher than 45 to 50 mm Hg often reflect true elevations in pulmonary artery pressures, as determined at right heart catheterization. In addition, about 20% to 30% of the population does not have tricuspid regurgitation,9 making it difficult to assess RVSP.
Currently, 6 treatments for PAH in SSc are FDA-approved.10 Actively considering the diagnosis and pressing for evaluation and documentation is important. Because the evaluation and management of PAH in SSc can be quite complex, the patient should be referred to physicians familiar with such management, such as pulmonologists, cardiologists, and rheumatologists. An echocardiogram should be included in the evaluation of any patient who has SSc and reports new or increasing dyspnea.
6. When is hypertension an emergency in SSc?
Essential hypertension develops in about 10% to 15% of patients with SSc.11 This condition requires treatment but does not constitute an emergency. A potentially catastrophic event, "renal crisis," occurs in about 15% to 20% of patients with diffuse cutaneous SSc and 2% to 3% of patients with limited cutaneous SSc.12,13 This is an emergency.
Essential hypertension tends to be asymptomatic, with little acute loss of renal function, at least in its early months or years. However, renal crisis tends to appear acutely: blood pressure levels are normal or even low 1 week before, and higher than 140/90 mm Hg (often near 160 to 200/100 mm Hg to 110 mm Hg or higher) the next. Other symptoms that tend to be prominent include new-onset headaches; a new stroke or neurological loss; chest pressure; shortness of breath, even at rest; and a recent increase in skin swelling and thickening. Laboratory tests may show a rising or elevated serum creatinine level, a new anemia (often hemolytic anemia, with fractured red blood cells, or schistocytes, on examination of the peripheral smear), a new thrombocytopenia (platelet count less than 100,000/µL), or proteinuria on urine dipstick. Urine volume may drop quickly to near anuric or oliguric levels.
The simplest way to think about renal crisis is to remember the following simple equation:
SSc + elevated blood pressure + rising creatinine level = RENAL CRISIS
Because patients may suddenly go into life-threatening arrhythmias, congestive heart failure, acute respiratory distress syndrome, and renal failure that requires dialysis, hospitalizing them and immediately starting treatment with an angiotensin-converting enzyme (ACE) inhibitor is prudent. ACE inhibitors are the only class of antihypertensives that has been shown to be lifesaving in renal crisis: without an ACE inhibitor, the patient has less than a 20% chance of living 6 to 12 months; with an ACE inhibitor, the survival rate is about 75% at 1 year, and patients who live 1 year usually will live more than 5 years.12
The dosage of the ACE inhibitor must be raised agressively to maximum levels within 1 to 3 days after starting treatment; other antihypertensives may be added, such as calcium channel blockers, angiotensin receptor blockers, and clonidine. The goal is to reduce the patient's blood pressure to 120/80 mm Hg or lower as soon as possible. Even so, the patient's serum creatinine level may rise for several weeks before it plateaus and declines; thus, the ACE inhibitor must be continued regardless of the rising creatinine level. About 40% to 50% of patients in whom renal crisis develops receive dialysis; within the following 18 to 24 months, between one-third and one-half are able to come off dialysis for the rest of their lives.12,13
There is no way to predict or prevent the onset of renal crisis. The best way to pick up renal crisis early is to have the patient take his blood pressure at home 3 times a week for screening. With any reading higher than 140/90 mm Hg, the test should be repeated later in the day. If the pressure is still elevated, the patient needs to be evaluated emergently and treatment with an ACE inhibitor needs to be started.
Checking the patient's serum creatinine level and starting ACE inhibitor therapy are prudent measures that must be taken emergently. If the hypertension appears to be associated with a rising creatinine level, hospitalizing the patient and administering an ACE inhibitor is the most prudent course. Also recommended is consulting with a nephrologist and a rheumatologist familiar with SSc.
7. When should I suspect heart involvement in SSc?
Significant heart involvement in SSc is almost exclusively a problem of patients with diffuse SSc (about 10% of these patients). Heart involvements include myocardial fibrosis that may result in dilated cardiomyopathy, pericardial effusions, and arrhythmias; the arrhythmias often are related to conduction disturbances resulting from scarring of areas around the conduction system.14
In patients in whom renal crisis occurs, an element of "heart failure" often develops that is related to the characteristic high angiotensin storm. ACE inhibitors often alleviate the heart failure and allow heart function to return to near normal.14
A myocardiopathy develops in other patients, resulting in diastolic dysfunction that is best managed with ACE inhibitors and diuretics. Palpitations and supraventricular arrhythmias are common in scleroderma, but the diagnosis often is not made because they cause the patient few significant problems.14
Ventricular arrhythmias may develop that require defibrillation or an implanted pacemaker and defibrillator. More often, the arrhythmia is supraventricular and may require long-term medication once the diagnosis is made (a 24-hour continuous Holter monitor may be used to document the type and severity of the arrhythmias).

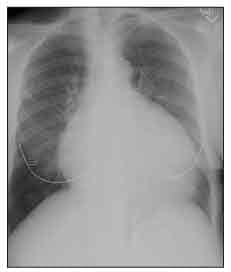
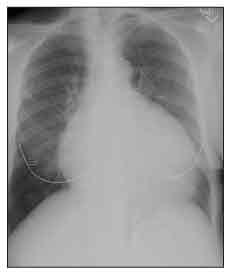
Figure 4 – Cardiomegaly is seen on this chest radiograph of a patient with systemic sclerosis (SSc). Although this enlarged heart shadow can be the result of SSc-related dilated cardiomyopathy, in this case, the patient had a large pericardial effusion.
Moderate or large pericardial effusions often are picked up accidentally on a chest x-ray film (Figure 4) or echocardiogram. Most are not associated with tamponade, but about 50% of patients with moderate or large pericardial effusions are at risk for renal crisis within the following 6 months.14
8. How much significance should I give to the complaint of heartburn in SSc?
Weakening of smooth muscle peristalsis of the lower esophagus and loss of sphincter tone in the gastroesophageal sphincter develop in at least 80% of patients with SSc; more than half also may have gastric hypomotility, resulting in gastric retention.15 Esophageal involvement usually is manifested by frequent heartburn that often is resistant to normal antireflux measures; it also may be associated with dysphagia and with regurgitation or vomiting of food that was recently ingested.
Gastric involvement (gastroparesis) may be manifested by early satiety (the patient feels full after only a few bites of food), abdominal bloating or abdominal swelling after eating, aggravation of heartburn, and regurgitation or vomiting of food that was recently ingested. Two major causes of weight loss and poor ability to gain weight are gastroparesis and small-intestine bacterial overgrowth (SIBO), discussed below.
Heartburn is the most common of these symptoms. Gastroesophageal reflux disease (GERD) with heartburn can be a big problem for patients with SSc. Although the heartburn of GERD in SSc may be resistant to simple antireflux measures, these measures should be instituted. They include elevating the head of the bed at night, eating frequent small meals, avoiding large meals after 6 pm, avoiding spicy foods, and avoiding tight-fitting abdominal garments and bending over at the waist. Proton pump inhibitors usually are required to suppress the hyperacidity in patients with SSc.
9. Are there other GI complaints in SSc I should be aware of?
Gastric antral vascular ectasia (GAVE) is an important cause of iron deficiency anemia in SSc that affects about 10% to 15% of patients. Because its characteristic appearance on upper endoscopy resembles the stripes of a watermelon, GAVE is also termed "watermelon stomach."
Hypomotility of smooth muscle in the small and large intestines also occurs frequently in SSc. Decreased peristalsis in the small intestine may lead to SIBO, with chronic diarrhea and abdominal bloating and distention.16 In fact, SIBO is the most common cause of chronic diarrhea in SSc.
For any patient with SSc who reports chronic diarrhea or bouts of diarrhea, examine stools for bacteria, Clostridium difficile, white blood cells, and ova and parasites. If the results are negative, the next moves are the following:
Obtain a lactulose hydrogen breath test (an early increase in breath hydrogen level suggests SIBO).
Have the patient avoid or minimize intake of milk products (because of any possible latent lactose intolerance).
Treat the patient with a broad-spectrum antibiotic, and plan to treat him with more than 1 antibiotic in sequence (eg, first ciprofloxacin, then metronidazole, then doxycycline, then amoxicillin; repeat the cycle with the antibiotics that were effective [alleviated symptoms]) for 10 to 14 days at a time.16,17 Rifaximin may be added to the list of antibiotics but is much more expensive than the others.17
The end points are a decrease in the number of stools per day and less abdominal bloating and distention. If the patient improves with antibiotics, you have the diagnosis. Once the patient has been through all 4 or 5 antibiotics, you will know which ones work and which do not. The patient can then be given a rotating schedule for taking effective antibiotics and, if need be, on a regular basis. Patients who do not improve with this approach should be referred for further evaluation to a gastroenterologist experienced in caring for patients with SSc.
In some patients, pseudo-obstruction of the small intestine becomes the major problem. The symptoms include marked, painful abdominal distention with absent bowel movements and absent flatus production. This condition requires the care of a gastroenterologist who can evaluate the patient and, if needed, prescribe nasogastric suction, bowel rest, and total parenteral nutrition (TPN). Some patients may require ambulatory TPN at home to survive the total lack of intestinal movement.
Decreased motility in the patient's large bowel often manifests as constipation and, much less often, as pseudo-obstruction. One reasonably successful treatment for constipation is polyethylene glycol 3350 (1 tablespoonful in an 8-oz glass of water taken once or twice a day). Again, pseudo-obstruction requires the care of a knowledgeable gastroenterologist.
CONCLUSIONS
Organ function (particularly heart, lung, and kidney) should be assessed frequently in the first 3 to 5 years of SSc. Even in the absence of clear-cut organ involvement, patients with early diffuse cutaneous SSc take their blood pressure 3 times a week, have their FVC and Dlco checked every 6 months to assess lung function, and obtain a 2-dimensional echocardiogram every year for the first 3 to 5 years of SSc. Patients with limited cutaneous SSc (who are at risk for ILD but are less likely to have significant renal or cardiac involvement) should have their FVC and DLCO checked every 6 months for the first 3 to 5 years of SSc. Testing for involvement of other organs should be performed as mandated by symptoms.
Discussion of SSc with patients should include where they are in the course of the disease and what they can do to help manage it. Reliable sources of pertinent patient information include the Scleroderma Foundation (http://www.scleroderma.org) and the International Scleroderma Network (http://www.sclero.org).
The good news is that many treatment strategies may help contain and modify the involvements that can occur in SSc. Uncovering the organ involvements as soon as possible, so that treatment can be instituted early, is paramount.
References:
References1. Mayes MD, Reveille JD. Epidemiology, demographics, and genetics. In: Clements PJ, Furst DE, eds. Systemic Sclerosis. 2nd ed. New York: Lippincott Williams & Wilkins; 2004:1-15.
2. Medsger TA. Classification, purpose. In: Clements PJ, Furst DE, eds. Systemic Sclerosis. 2nd ed. New York: Lippincott Williams & Wilkins; 2004:17-29.
3. Steen VD, Powell DL, Medsger TA Jr. Clinical correlations and prognosis based on serum autoantibodies in patients with systemic sclerosis. Arthritis Rheum. 1988;31:196-203.
4. Steen VD. The many faces of scleroderma. Rheum Dis Clin North Am. 2008;34:1-15, v.
5. Steen VD, Conte C, Owens GR, Medsger TA Jr. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994;37:1283-1289.
6. Au K, Khanna D, Clements PJ, et al. Current concepts in disease-modifying therapy for systemic sclerosis-associated interstitial lung disease: lessons from clinical trials. Curr Rheumatol Rep. 2009;11:111-119.
7. Williams MH, Das C, Handler CE, et al. Systemic sclerosis associated pulmonary hypertension: improved survival in the current era. Heart. 2006;92:926-932.
8. Arcasoy SM, Christie JD, Ferrari VA, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003;167:735-740.
9. Merkel PA, Clements PJ, Reveille JD, et al; OMERACT 6. Current status of outcome measure development for clinical trials in systemic sclerosis: report from OMERACT 6. J Rheumatol. 2003;30:1630-1647.
10. Rubin LJ. Treatment of pulmonary arterial hypertension due to scleroderma: challenges for the future. Rheum Dis Clin North Am. 2008;34:191-197, viii.
11. Steen VD, Syzd A, Johnson JP, et al. Kidney disease other than renal crisis in patients with diffuse scleroderma. J Rheumatol. 2005;32:649-655.
12. Steen VD. Scleroderma renal crisis. Rheum Dis Clin North Am. 2003;29:315-333.
13. Penn H, Denton CP. Diagnosis, management and prevention of scleroderma renal disease. Curr Opin Rheumatol.2008;20:692-696.
14. Follansbee WP, Marroquin OC. Cardiac involvement in systemic sclerosis. In: Clements PJ, Furst DE,eds. Systemic Sclerosis. 2nd ed. New York: Lippincott Williams & Wilkins; 2004:195-220.
15. Domsic R, Fasanella K, Bielefeldt K. Gastrointestinal manifestations of systemic sclerosis. Dig Dis Sci. 2008;53:1163-1174.
16. Kowal-Bielecka O, Landewé R, Avouac J, et al; EUSTAR Co-Authors. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR). Ann Rheum Dis. 2009;68:620-628.
17. Parodi A, Sessarego M, Greco A, et al. Small intestinal bacterial overgrowth in patients suffering from scleroderma: clinical effectiveness of its eradication. Am J Gastroenterol. 2008;103:1257-1262.














