Identifying and managing pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) may lead to increased pulmonary vascular resistance, compromised vasoreactivity, right heart failure, and death. PAH, a subset of pulmonary hypertension (PH), classically is associated with systemic sclerosis but also occurs with other rheumatologic conditions. Early diagnostic strategies are essential. The symptoms of PAH often are nonspecific; the most common presenting symptom is dyspnea.
Pulmonary arterial hypertension (PAH), a progressive disease, may lead to increased pulmonary vascular resistance, compromised vasoreactivity and, eventually, right-sided heart failure and death. PAH is a subset of pulmonary hypertension (PH) and a hemodynamic observation. Based on reports at a recent conference, PH is defined as a resting mean pulmonary artery pressure (PAP) greater than 25 mm Hg; PAH is defined as PH in addition to pulmonary capillary wedge pressure (PCWP) 15 mm Hg or greater (the absence of elevated PCWP rules out the presence of left-sided heart disease as the cause of PH).1 PAH is a well-documented and increasingly appreciated cause of morbidity and mortality.2 Although classically associated with systemic sclerosis (SSc), this condition also occurs in the setting of systemic lupus erythematosus (SLE), mixed connective-tissue disease (MCTD) and, less often, rheumatoid arthritis, Sjgren syndrome, and inflammatory myositis.
Because the diagnosis of PAH often is made years after symptom onset,3 and hemodynamic severity correlates with subsequent mortality, early diagnostic strategies are essential. Physicians need to maintain a high index of suspicion to achieve early diagnosis. Right heart catheterization (RHC) currently is the only modality used to differentiate PAH from PH and make a definitive diagnosis.
In this article, we describe the prevalence, pathophysiology, and approaches to the diagnosis of PAH. We also offer guidelines for the management of PAH and the management of PH.
PREVALENCE
Connective-tissue disease (CTD)-associated PAH (CTD-PAH) is a rare disease; the estimated prevalence of resting CTD-PAH is 2.3 to 10 cases per million persons.4 Overall, the reported prevalence of PAH is 5% to 50% in patients with SSc,5 21% to 29% in those with MCTD, and 5% to 43% in those with SLE.6 The wide range in reported frequency of PAH probably is the result of differences in the methods and criteria used to make a diagnosis, as well as differences in patient populations.
The prevalence of SSc-PAH, as indicated by transthoracic echocardiography, was as high as 35%.7 The prevalence of resting PAH, based on cardiac catheterization (the gold standard), was 12% with concomitant interstitial lung disease (ILD) and 8% without ILD.5 However, autopsy studies report evidence of pulmonary vasculopathy in 50% to 70% of patients with limited scleroderma, suggesting discordance between hemodynamic and histopathological diagnoses.
PATHOPHYSIOLOGY
Pathological mechanisms
Several pathophysiological mechanisms lead to elevated pulmonary vascular resistance in patients with CTD-associated PAH. The process of pulmonary vascular disease is accompanied by endothelial dysfunction, activation of fibroblasts and smooth muscle cells, cross talk between cells within the vascular wall, and recruitment of progenitor cells. Endothelial dysfunction in PAH is reflected by reduced production of vasodilators (eg, nitric oxide and prostacyclin) and increased production of vasoconstrictors (eg, endothelin-1). Because CTD is considered a subgroup belonging to the disease entity of PAH,2 the pathological anatomy of pulmonary vessels has been regarded as similar to the vascular changes found in other forms of PAH.8

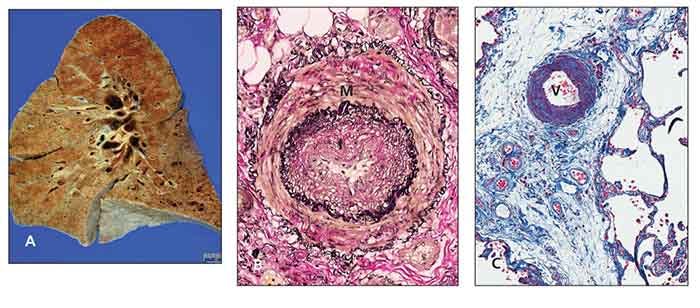
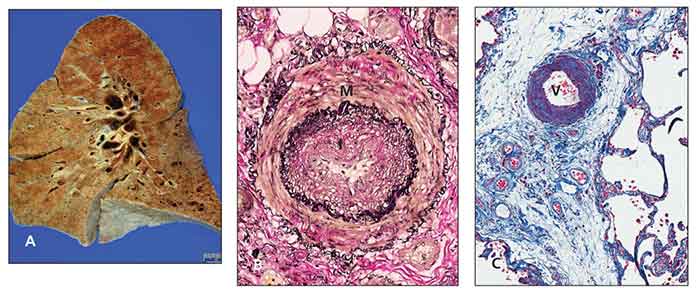
Figure – Pathology slides show scleroderma lung and pulmonary arterial hypertension. The gross appearance with lower lobe fibrosis and honeycomb change is identical to that of idiopathic pulmonary fibrosis (A). An artery has medial hypertrophy (M) and intimal hyperplasia (I) (Elastica van Gieson stain, original magnification ×100) (B). A vein (V) in the interlobular septum shows marked thickening resulting from intimal hyperplasia (trichrome stain, original magnification ×100) (C). (Images courtesy of Dr Michael Fishbein.)
Regardless of the cause, typical pulmonary arteriopathy includes medial hypertrophy of muscular and elastic arteries, dilation and intimal atheromas of elastic pulmonary arteries, and thrombotic microangiopathy (Figure).9 However, differences in clinical behavior and response to therapy between CTD-PAH and other forms of PAH are evident.10 This finding may be explained by coinciding patterns of arteriolar disease and a pulmonary veno-occlusive disease–like pattern in CTD populations.11,12 In addition, the absence of plexiform lesions in SSc-PAH, a classic pathological feature observed in idiopathic PAH, was noted in a recent autopsy series.12 In summary, PAH is a complex process characterized by an imbalance among vasodilators and vasoconstrictors, growth inhibitors and mitogenic factors, and an inflammatory/proliferative vasculopathy that probably results from endothelial cell dysfunction or injury.
Immunological mechanisms
These mechanisms also may contribute to the development of PAH, especially given the autoimmune nature of the underlying CTD disorder. Although no single antibody or combination of autoantibodies predicts the development of PAH, several studies have reported an association between the presence of particular antibodies and PH.
Although direct injury mediated by immunological factors has not yet been reported clearly, the presence of circulating antinuclear antibodies, IgG deposition, rheumatoid factor, and other inflammatory mediators has been identified in the pulmonary vasculature of patients with CTD-PAH. Several groups also have reported an association with elevated levels of platelet-derived growth factor, RANTES/CCL5, and fractalkine (CX3CL1) in pulmonary arteries of patients with advanced PAH.13 In addition, the presence of anti-U1RNP, anticentromere, antinucleolar, and anticardiolipin antibodies has been appreciated with CTD-associated PAH.
These and other studies suggest an immunological insult causing PH in some patients with CTD.14 This finding may explain the hemodynamic improvement seen in some patients who have CTD and are treated with immunosuppressive therapy, typically the patients who have SLE and not the majority of patients.15
DIAGNOSIS

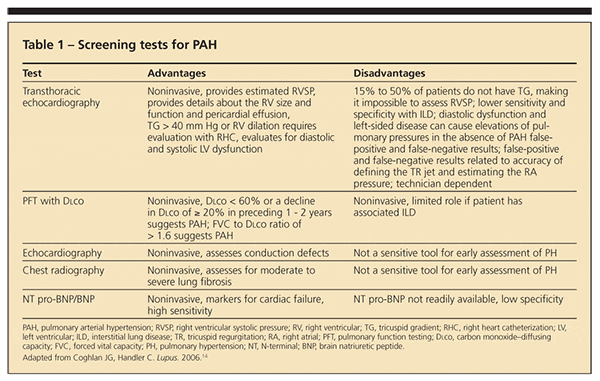
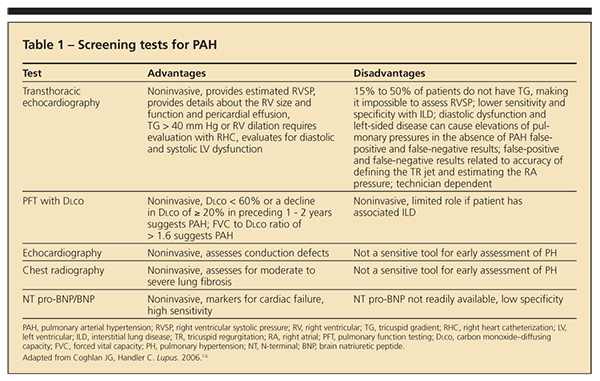
In 2008, the 4th World Symposium on Pulmonary Hypertension defined 5 categories in which diseases were grouped according to specific therapeutic interventions directed at dealing with the cause: (1) PAH (including CTD), (2) PH with left-sided heart disease, (3) PH associated with lung disease or hypoxemia or both, (4) PH resulting from chronic thrombotic or embolic disease or both, and (5) miscellaneous.16 Within each category are subsets that reflect diverse causes and sites of injury. Table 1 describes the available screening tests for PAH and their advantages and disadvantages in helping clinicians achieve early diagnosis.
Symptoms and physical examination
The symptoms of PAH often are nonspecific.3 The most common presenting symptom is dyspnea (seen in 60% of patients). Other symptoms, which increase during the disease course, include fatigue (73% of patients), chest pain (47%), syncope (36%), and edema (37%). The probability of PAH occurring is increased when the physical examination shows signs of right-sided heart strain or failure, such as increased P2, prominent jugular venous pulsations, right-sided S3 or S4 or both, holosystolic murmur of tricuspid regurgitation, and ascites and edema. Keep in mind that symptoms of right-sided heart involvement may occur late in the course of PAH.
Electrocardiography
The ECG findings of PAH reflect right-heart-chamber adaptations to increased pulmonary vascular resistance. Right axis deviation (79% of cases) and right ventricular (RV) hypertrophy (87% of cases) are common in severe PAH.3 However, electrocardiography is thought to be inadequate as a single test in screening to rule out clinically relevant PAH.
Chest radiography
Obtaining a chest radiograph is necessary in the diagnostic evaluation of PAH. Like electrocardiography, however, chest radiography lacks sufficient sensitivity to function as a screening test for PAH. In the setting of PH, a chest radiograph may feature enlarged main pulmonary and hilar artery shadows; peripheral "pruning" of vessels; loss of the retrosternal air space (lateral view), suggesting RV enlargement; increased radiological index of PAH; and increased right (more than 16 mm) or left (more than 18 mm) descending pulmonary artery caliber.
Pulmonary function testing
Several studies have shown that up to 50% of patients with PAH have a mild spirometric restriction with a moderate reduction in carbon monoxide–diffusing capacity (DLCO). In particular, SSc represents a unique population of patients who are screened for PAH and ILD (ie, fibrosis) routinely, given their strong association with subsequent mortality. In patients who have SSc, a decreased or decreasing DLCO less than 55% of predicted17,18 or increased forced vital capacity (FVC):DLCO ratio greater than 1.6 can predict the development or progression of PAH.19 However, clinicians should not rely on these tests to exclude patients where the pretest probability of PAH occurring is high.
If indicated, a high-resolution CT scan of the chest may be needed to evaluate the degree of fibrosis. Typically, patients with an FVC less than 60% or an FVC 60% to 70% associated with grade 3 or 4 fibrosis are classified as having ILD associated with PH.
Serology
No specific blood test is diagnostic of PAH. However, a serological evaluation helps characterize whether the PAH is idiopathic or associated with another World Health Organization (WHO) group 1 condition.20 Biomarkers that are prognostic predictors of established PAH include brain natriuretic peptide (BNP), N-terminal pro-BNP, uric acid, anticentromere antibodies, antinucleolar antibodies, and anticardiolipin antibodies.
Doppler echocardiography
This modality currently is considered the noninvasive screening test of choice for evaluating PH. The WHO recommends baseline and then yearly Doppler echocardiography (DE) evaluation for patients with SSc.
The sensitivity and specificity of DE for estimating PAP are 0.79 to 1.0 and 0.68 to 0.98, respectively. DE estimation of systolic PAP requires a detectable and adequate tricuspid jet velocity and an assessment of right atrial (RA) pressure by inferior vena cava collapsibility, a standardized value (5 or 10 mm Hg), or a clinical estimation of jugular venous pressure. With use of the maximum velocity (V) of the tricuspid regurgitant (TR) jet, the systolic pressure gradient between the RV and RA is calculated by the modified Bernoulli equation (4V2); the sum of this pressure gradient and the RA pressure estimates pulmonary artery systolic pressure, assuming no obstruction to RV blood flow.21 In about 15% to 30% of patients, TR jet is not detectable. Other DE signs include pericardial effusion, RA enlargement, and RV strain.
Echocardiography as a screening tool
In specialized centers where there is interest in PH, DE can evaluate the degree of PH, prognosticating variables (RA and RV enlargement, pericardial effusion, RV ejection fraction), and other potential causes of PH (left ventricular systolic/diastolic dysfunction, valvular heart disease, and intracardiac shunting [with bubble study]). With careful study, DE also may be used to document the presence and degree of diastolic dysfunction and might help distinguish PAH from venous hypertension.
In real-life practice, DE is associated with a high rate of false-positive studies (in the presence of ILD) and false-negative studies (early PAH). Therefore, DE is not an acceptable replacement for RHC, especially because nonsimultaneous assessments have been shown to be statistically inaccurate in a population in which the prevalence of PH is high.22 Of note, this correlation is weakened substantially in the setting of parenchymal lung disease (WHO class 3 PAH), especially ILD. Patients with elevated estimated RV systolic pressure (higher than 35 mm Hg) or evidence of right-sided strain on an ECG, FVC:DLCO ratio higher than 1.6, or a decline in DLCO of 15% or more in the previous 1 to 2 years should be referred for RHC evaluation.
The Scleroderma Pulmonary Hypertension Quality Enhancement Research Initiative, an observational cohort designed to improve detection and management of SSc-PAH, showed that 84% of rheumatologists are using DE for patients with SSc. However, only 48% of the DE results reported tricuspid regurgitation, emphasizing that physicians should ask specifically for assessment of the right side of the heart in a patient in whom PAH is suspected.
Right heart catheterization
RHC is the confirmatory test for PH. It allows for precise establishment of the diagnosis and type of PH, disease severity, prognosticating hemodynamic data, and pulmonary vascular reserve. PAH is defined as resting mean PAP higher than 25 mm Hg (or higher than 30 mm Hg with exercise), with a PCWP lower than 15 mm Hg.23 In PH centers, the risk of serious adverse events attributable to the procedure is quite low.
TREATMENT
General guidelines
PH is best managed by a multidisciplinary team of physicians (pulmonologists, cardiologists, and rheumatologists) who are experienced in treating patients with this condition. Primary care and other comanaging physicians should recommend regular, cautious, supervised exercise and ask patients to "listen to their body." Patients should be counseled to stop smoking. Prophylaxis for pulmonary infections (influenza and pneumococcal vaccination) should be administered. Because persistent hypoxemia may accelerate the course of PAH, aggressive evaluation should be performed for anemia and to maintain oxygen saturation at higher than 90% at rest and during supervised exercise and sleep.
Summary of medical therapy
Seven medications are FDA-approved for PAH therapy (Table 2). Endothelin and phosphodiesterase (PDE)-5 blockers are given as oral agents; prostacyclin derivatives are approved for intravenous or subcutaneous administration. Treatment of patients with PAH should be individualized according to the severity of functional impairment. Intravenous prostacycli

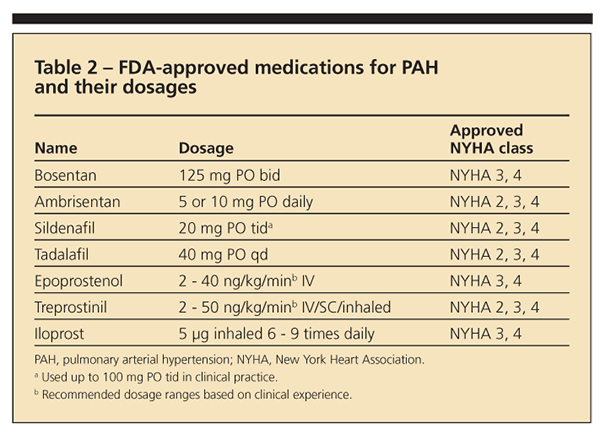
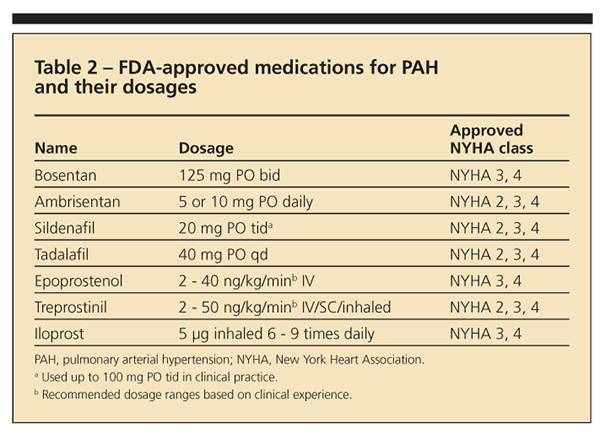
n therapies have been best studied for severe PAH (particularly in patients in WHO class 4 or those with right-sided heart failure) and is the gold standard in this context.24,25 However, treatment with oral endothelin-A receptor antagonist (ETRA) or PDE-5i is reasonable for patients in WHO class 2 or 3 (and select patients in class 4).24,26
Given the multiple mechanisms of action, there is a scientific rationale for the use of combination therapy for PAH, and such therapy shows promise. Treatment for patients with less severe PAH (eg, those in WHO class 1 or those with exercise-induced disease) is controversial. Patients with WHO class 3 PAH who are unresponsive to treatment with ETRA or PDE-5i should be given additional therapy with parenteral prostanoids.24,26
Pharmacological management of PAH is exceptionally expensive, and current studies of "add-on" and "goal-directed" therapy show promise with these approaches.27,28 The effectiveness of therapy should be monitored by PAH centers using functional class and symptoms (eg, dyspnea index), exercise capacity (6-minute walk distance), echocardiography, and hemodynamics.
Managing PH
The management of PH (resulting from left-sided heart disease, chronic thromboembolic disorder, lung disease associated with hypoxemia, or miscellaneous disorders) is based on the underlying cause. However, clinical trials of bosentan and epoprostenol for systolic heart failure were terminated early because of increased hospitalizations of participants for heart failure or death.29,30 Therefore, PAH-specific therapies should not be used in patients with PH resulting from left-sided heart disease (typically manifested by PCWP greater than 15 mm Hg on RHC). Examples of left-sided heart disease include systolic and diastolic dysfunction; valvular diseases may produce an increase in left atrial pressure, with passive backward transmission of the pressure leading to increased PAP. In this situation, the pulmonary vascular resistance is normal (less than 3.0 Wood units) and there is no gradient between mean PAP and PCWP (transpulmonary gradient lower than 12 mm Hg).
References:
References
1.
Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension.
J Am Coll Cardiol
. 2009;54(suppl 1):S55-S66.
2.
Simonneau G, Galiè N, Rubin LJ, et al. Clinical classification of pulmonary hypertension.
J Am Coll Cardiol
. 2004;43(12 suppl S):S5-S12.
3.
Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension: a national prospective study.
Ann Intern Med
. 1987;107:216-223.
4.
Condliffe R, Kiely DG, Peacock AJ, et al. Connective tissue diseaseâassociated pulmonary arterial hypertension in the modern treatment era.
Am J Respir Crit Care Med
. 2009;179:151-157.
5.
Mukerjee D, St George D, Coleiro B, et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach.
Ann Rheum Dis
. 2003;62:1088-1093.
6.
Winslow TM, Ossipov MA, Fazio GP, et al. Five-year follow-up study of the prevalence and progression of pulmonary hypertension in systemic lupus erythematosus.
Am Heart J
. 1995;129:510-515.
7.
Wigley FM, Lima JA, Mayes M, et al. The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study).
Arthritis Rheum
. 2005;52:2125-2132.
8.
Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection.
Hum Pathol
. 1997;28:434-442.
9.
Pietra GG, Capron F, Stewart S, et al. Pathologic assessment of vasculopathies in pulmonary hypertension.
J Am Coll Cardiol.
2004;43(12 suppl S):S25-S32.
10.
Fisher MR, Mathai SC, Champion HC, et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension.
Arthritis Rheum.
2006;54:3043-3050.
11.
Dorfmüller P, Humbert M, Perros F, et al. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases.
Hum Pathol.
2007;38:893-902.
12.
Overbeek MJ, Vonk MC, Boonstra A, et al. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: a distinctive vasculopathy.
Eur Respir J
. 2009;34:371-379.
13.
Balabanian K, Foussat A, Dorfmüller P, et al. CX(3)C chemokine fractalkine in pulmonary arterial hypertension.
Am J Respir Crit Care Med
. 2002;165:1419-1425.
14.
Coghlan JG, Handler C. Connective tissue associated pulmonary arterial hypertension.
Lupus
. 2006;15:138-142.
15.
Sanchez O, Sitbon O, Jaïs X, et al. Immunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertension.
Chest
. 2006;130:182-189.
16.
McLaughlin VV, Archer SL, Badesch DB, et al; American College of Cardiology Foundation Task Force on Expert Consensus Documents; American Heart Association; American College of Chest Physicians; American Thoracic Society, Inc; Pulmonary Hypertension Association. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association.
J Am Coll Cardiol
. 2009;53:1573-1619.
17.
Launay D, Mouthon L, Hachulla E, et al. Prevalence and characteristics of moderate to severe pulmonary hypertension in systemic sclerosis with and without interstitial lung disease.
J Rheumatol
. 2007;34:1005-1011.
18.
Steen V, Medsger TA Jr. Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement.
Arthritis Rheum
. 2003;48:516-522.
19.
Steen VD, Graham G, Conte C, et al. Isolated diffusing capacity reduction in systemic sclerosis.
Arthritis Rheum
. 1992;35:765-770.
20.
McGoon M, Gutterman D, Steen V, et al; American College of Chest Physicians. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines.
Chest
. 2004;126(1 suppl):S14-S34.
21.
Yock PG, Popp RL. Noninvasive estimation of right ventricular systolic pressure by Doppler ultrasound in patients with tricuspid regurgitation.
Circulation
. 1984;70:657-662.
22.
Selimovic N, Rundqvist B, Bergh CH, et al. Assessment of pulmonary vascular resistance by Doppler echocardiography in patients with pulmonary arterial hypertension.
J Heart Lung Transplant
. 2007;26:927-934.
23.
Rubin LJ; American College of Chest Physicians. Diagnosis and management of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines.
Chest
. 2004;126(1 suppl):S7-S10.
24.
Hoeper MM, Galiè N, Simonneau G, Rubin LJ. New treatments for pulmonary arterial hypertension.
Am J Respir Crit Care Med
. 2002;165:1209-1216.
25.
Strauss WL, Edelman JD. Prostanoid therapy for pulmonary arterial hypertension.
Clin Chest Med
. 2007;28:127-142; ix.
26.
Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension.
N Engl J Med
. 2004;351:1425-1436.
27.
Hoeper MM, Barberà JA, Channick RN, et al. Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension.
J Am Coll Cardiol
. 2009;54(suppl 1):S85-S96.
28.
Hoeper MM, Markevych I, Spiekerkoetter E, et al. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension.
Eur Respir J
. 2005;26: 858-863.
29.
Kaluski E, Cotter G, Leitman M, et al. Clinical and hemodynamic effects of bosentan dose optimization in symptomatic heart failure patients with severe systolic dysfunction, associated with secondary pulmonary hypertension-a multi-center randomized study.
Cardiology
. 2008;109:273-280.
30.
Califf RM, Adams KF, McKenna WJ, et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: The Flolan International Randomized Survival Trial (FIRST).
Am Heart J.
1997;134:44-54.














