Inflammatory bone loss in joint replacements: The key mediators
The immune system plays a dynamic role in physiological and pathogenic bone remodeling. Macrophage-, stromal-, and lymphocyte-derived cytokines may induce the activation or inhibition of bone cells and their precursors and cause bone deposition or bone resorption.
The dynamic role that the immune system plays in physiological and pathogenic bone remodeling is well established. Macrophage-, stromal-, and lymphocyte-derived cytokines may induce the activation or inhibition of bone cells and their precursors and cause bone deposition or bone resorption.1,2 Although progress has been made in understanding the immune and bone systems in the field of osteoimmunology, understanding how inflammatory responses triggered by orthopedic implant wear directly or indirectly impact osteolysis remains a complex and often convoluted area of study.
This 2-part article offers current insights into inflammatory bone loss in joint replacements. In the
first part, “Inflammatory bone loss in joint replacements: The mechanisms” (The Journal ofMusculoskeletal Medicine, June 2010, page 209), we described the intracellular mechanisms involved in implant debris–induced osteolysis and new avenues for pharmacological treatment.
This second part elaborates on the osteoimmunological science.
Proinflammatory mediators
In vitro experimentation and in vivo animal models of osteolysis have shown that key proinflammatory mediators, such as receptor activator of nuclear factor kB ligand (RANKL), interleukin (IL)-1b, IL-6, tumor necrosis factor α (TNF-α), cyclooxygenase (COX)-2, and prostaglandin E2 (PGE2), play a significant role in osteoclast precursor recruitment, differentiation, and activation.1,3-6 For example, mice genetically deficient in RANKL (a key osteoclast activator) show evidence of abnormally increased bone formation or deficient bone resorption compared with normal mice.7,8 Also, mice genetically deficient in the inflammatory cytokines IL-1b and TNF-α or the specific receptors for these cytokines exhibit reduced particle-induced osteolysis in vitro and in vivo compared with their wild type counterparts.9,10
Clinical studies of wear debris–induced osteolysis have demonstrated that treatment with specific inflammatory blockers, such as selective inhibitors of COX-2, reduces particle-induced osteolysis by hindering PGE2 production.11 Similar results have been observed with TNF-α inhibitors and with IL-1 receptor antagonist (IL-1Ra), which keeps IL-1b from acting in a paracrine and autocrine manner on macrophages.
A retrovirus-induced overexpression of IL-1Ra in a mouse model showed significantly increased protection against particle-induced osteolysis compared with a group of mice that did not have overexpression of IL-1Ra.12 Similarly, in a clinical cohort of patients who had undergone a total hip arthroplasty, a genetic polymorphism that results in overexpression of IL-1Ra rendered them less likely to experience osteolysis.13 Conversely, a polymorphism resulting in overexpression of IL-6 in a different cohort of patients increased the risk of osteolysis in those who had orthopedic implants.13
Although correlations between specific inflammatory mediators and osteolysis have been found, the process is so multifactorial that isolating a specific mediator as the single determinant of inflammatory bone loss is virtually impossible. The overall balance between osteoclastogenic and antiosteoclastogenic factors purportedly dictates the fate of bone-to-implant integrity.
Promising treatments for patients with osteolysis include investigational monoclonal antibodies, such as one that targets RANKL (denosumab), which has been shown to be effective in the treatment of patients with osteoporosis.14 Because RANKL is a cytokine that is essential for the function of osteoclasts, inhibiting RANKL with denosumab counters inflammatory bone loss.15,16
The role of IL-17
Involvement of the adaptive immune system (T lymphocytes) in osteolysis induced by “everyday”-wear debris has been to some extent controversial. However, mounting evidence suggests that T cell–derived cytokines and costimulatory cell receptor molecules that are shared by both immune and bone systems (eg, CD80, CD86, CD54) may play key roles in the process of osteolysis.17,18
The role of IL-17 is of particular interest because studies have shown the significance of activated CD4+ T-lymphocyte secretion of IL-17 (by TH17 subsets) in combination with expression or secretion of RANKL to induce osteoclast differentiation.19 Takayanagi1 demonstrated that IL-17 can induce osteoclastogenesis by inducing RANKL expression on synovial fibroblasts. Other authors have shown the role of IL-17 and IL-23 (an inducer of IL-17) in collagen-induced arthritis (severe disease does not develop in knockout mice deficient in IL-17 or IL-23).20
The newfound role of IL-17 in osteoclastogenesis is important because in earlier studies, interferon-gamma– and IL-4–secreting CD4+ T lymphocytes were shown to inhibit osteoclast differentiation, despite their concomitant production of bone-resorbing RANKL. These earlier findings cast some doubt on the ability of T lymphocytes to contribute to the osteolytic process; however, the discovery of IL-17–producing T lymphocytes solidified concrete mechanisms by which T-cell–aided osteolysis can occur.
Some clinical evidence of the role of IL-17 in aseptic loosening of prosthetic joints has been described. In a cohort of 8 cases of revision surgery, immunohistochemical staining of peri-implant tissues demonstrated the presence of IL-17 in all samples.21 However, evidence for the involvement of IL-17 in implant debris–induced osteolysis remains limited and requires further investigation.
Molecular genesis of debris-induced inflammation
Several immune factors may influence bone remodeling, particularly in aberrant physiological conditions in which implant-generated debris (particles, ions) is continually challenging immune and stromal cells, inducing the release of inflammatory and bone resorbing osteoclastogenic cytokines. However, how and why immune and stromal cells sense, engulf, and respond to sterile nonpathogen materials such as implant debris, leading to an inflammatory response, is still unknown. At the cellular level, implant-generated debris clearly is engulfed and elicits proinflammatory cytokines from innate and adaptive immune cells. Still unclear is how this process takes place at the molecular level.
Some studies suggest that serum complement opsonization of implant debris is an important
factor that mediates particle uptake by phagocytes (macrophages). Complement proteins, immunoglobulins, and other cell surface receptors have been shown to preferentially bind to
implant debris and facilitate phagocytosis.22,23
Complement receptor CR3 and opsonin-independent scavenger receptor MARCO were shown to be necessary to trigger a macrophage inflammatory response to polymethylmethacrylate (PMMA) particles and to titanium particles, respectively.24,25 Other studies also suggest that toll-like receptors, which are innate immune system receptors for pathogen-associated molecular patterns (PAMPs), play a role in particle-induced inflammation.26 Also influential is the protein coating (biofilm) around implant-generated particles, which interacts with cell membrane receptors and can influence their uptake or bioreactivity/antigenicity.
Activation and phosphorylation
What is known of the intracellular mechanisms involved in implant debris–induced inflammatory responses is that several debris types (eg, PMMA and titanium particles) may induce the activation and phosphorylation of all major mitogen-activated protein kinase (MAPK) subgroups, including p38, extracellular signal–regulated kinases (ERKs), and c-Jun amino-terminal kinases (JNKs) in the cytosol of macrophages.27 Activation of these important intracellular signaling molecules has been linked to both proinflammatory responses in immune cells and direct osteoclastogenic effects in osteoclast precursors. The next generation of anti-inflammatory pharmaceuticals probably will focus on inhibiting these pathways in peri-implant immune cells (macrophages).

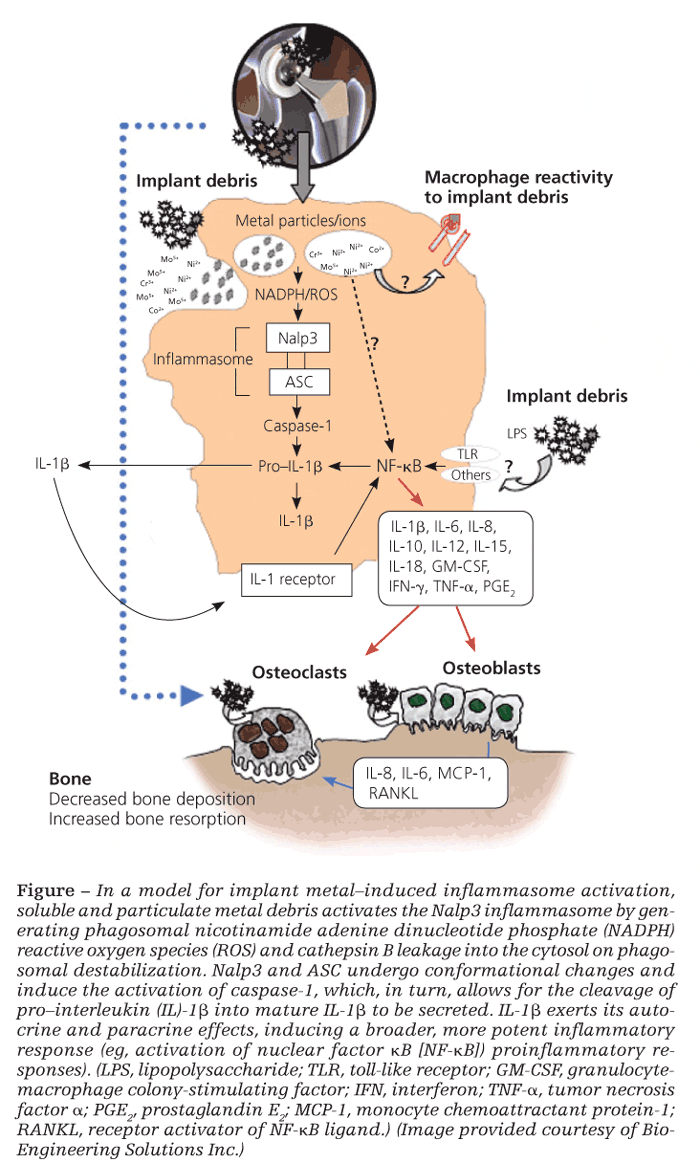
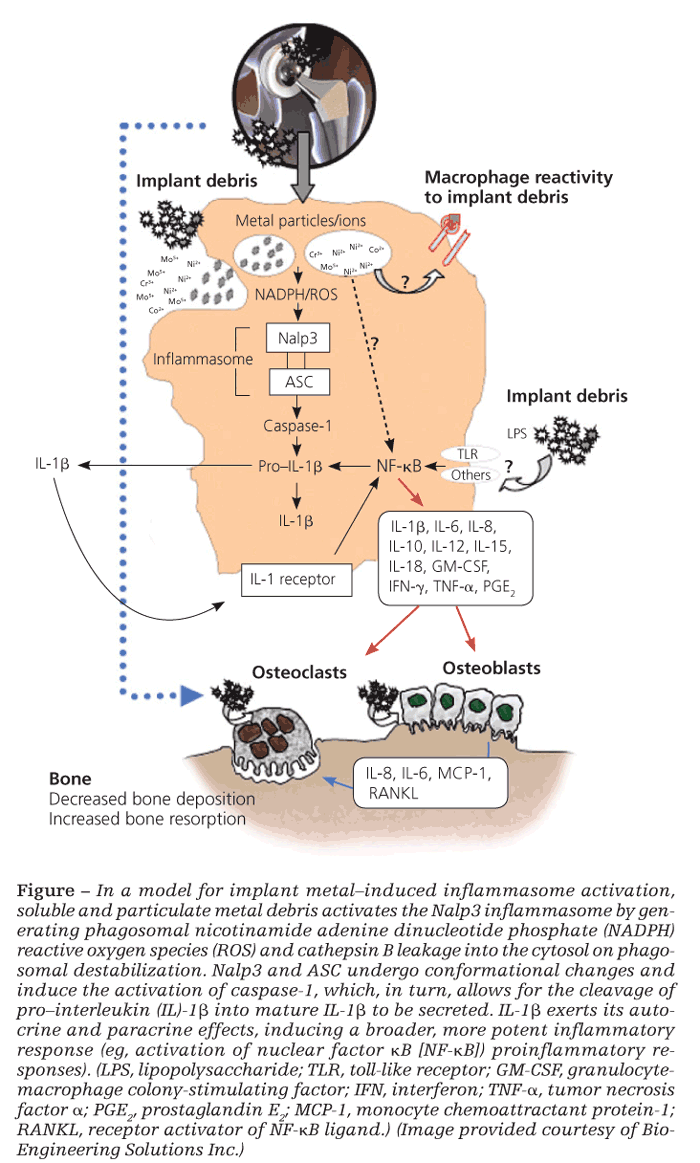
It is also well established that implant debris may efficiently activate transcription factors nuclear factor kB (NF-kB) and activator protein 1, leading to the production of proinflammatory cytokines TNF-α, IL-6, and PGE2, which also have important implications in the generation of osteoclasts.28 However, what has been confounding for several years is how relatively inert sterile debris can induce the activation of these intracellular inflammatory pathways that are otherwise activated via specific receptors for PAMPs found only in biological antigens (bacteria, viruses, fungi).
The inflammasome danger signaling pathway is one such recently discovered pathway. In it, nonbiological agents can trigger intracellular danger signals (eg, reactive oxygen species, adenosine triphosphate, cathepsin B) that are detected by NOD-like receptors in the cytosol of cells to induce a proinflammatory response (Figure).29,30
Danger-associated molecular patterns
Of the NOD-like receptors, the Nalp3 inflammasome specifically has been shown to respond to danger-associated molecular patterns elicited by nonbiological challenge agents-such as asbestos, alum, UV light, and implant debris-resulting in the secretion of proinflammatory IL-1b and the subsequent inflammatory response.30 Once stimulated, IL-1b can feed back and activate cells in both an autocrine and a paracrine fashion through the IL-1 receptor–MyD88 complex that leads to NF-kB translocation/activation and production of other “osteoclastic” proinflammatory cytokines (eg, IL-6 and TNF-α). Wear debris also may act independently of the inflammasome to activate NF-kB and p38, ERK, and JNK MAPK through other, still unknown mechanisms.
It is difficult to pinpoint which proinflammatory pathway is the key in each patient. In the context of implant debris–induced inflammation, however, the importance of the inflammasome danger-signaling pathway is underscored by findings that inflammasome-induced IL-1b is necessary to induce osteolysis and hypersensitivity responses in vivo.31
References:
References
1. Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292-304.
2. Takayanagi H. Inflammatory bone destruction and osteoimmunology. J Periodontal Res. 2005;40:287-293.
3. Bendre MS, Montague DC, Peery T, et al. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone. 2003;33:28-37.
4. Gao Y, Morita I, Maruo N, et al. Expression of IL-6 receptor and GP130 in mouse bone marrow cells during osteoclast differentiation. Bone. 1998;22:487-493.
5. Ma T, Miyanishi K, Suen A, et al. Human interleukin-1-induced murine osteoclastogenesis is dependent on RANKL, but independent of TNF-alpha. Cytokine. 2004;26:138-144.
6. Ma T, Miyanishi K, Trindade MC, et al. Interleukin 1 receptor antagonist inhibits localized bone formation in vivo. J Rheumatol. 2003;30:2547-2552.
7. Odgren PR, Kim N, MacKay CA, et al. The role of RANKL (TRANCE/TNFSF11), a tumor necrosis factor family member, in skeletal development: effects of gene knockout and transgenic rescue. Connect Tissue Res. 2003;(44, suppl 1):S264-S271.
8. Ren W, Wu B, Peng X, et al. Implant wear induces inflammation, but not osteoclastic bone resorption, in RANK(-/-) mice. J Orthop Res. 2006;24:1575-1586.
9. Boyce BF, Li P, Yao Z, et al. TNF-alpha and pathologic bone resorption. Keio J Med. 2005;54:127-131.
10. Wei S, Kitaura H, Zhou P, et al. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. 2005;115: 282-290.
11. Zhang X, Morham SG, Langenbach R, et al. Evidence for a direct role of cyclo-oxygenase 2 in implant wear debris-induced osteolysis. J Bone MinerRes. 2001;16:660-670.
12. Yang SY, Wu B, Mayton L, et al. Protective effects of IL-1Ra or vIL-10 gene transfer on a murine model of wear debris-induced osteolysis. Gene Ther. 2004;11:483-491.
13. Gordon A, Kiss-Toth E, Stockley I, et al. Polymorphisms in the interleukin-1 receptor antagonist and interleukin-6 genes affect risk of osteolysis in patients with total hip arthroplasty. Arthritis Rheum. 2008;58:3157-3165.
14. Lewiecki EM, Miller PD, McClung MR, et al; AMG 162 Bone Loss Study Group. Two-year treatment with denosumab (AMG 162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res. 2007;22:1832-1841.
15. Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis [published correction appears in N Engl J Med. 2009;361:1914]. N Engl J Med. 2009;361:756-765.
16. Miller PD, Bolognese MA, Lewiecki EM, et al; Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone. 2008;43:222-229.
17. Caicedo MS, Pennekamp PH, McAllister K, et al. Soluble ions more than particulate cobalt-alloy implant debris induce monocyte costimulatory molecule expression and release of proinflammatory cytokines critical to metal-induced lymphocyte reactivity. J Biomed Mater Res A. 2010;93:1312-1321.
18. Farber A, Chin R, Song Y, et al. Chronic antigen-specific immune-system activation may potentially be involved in the loosening of cemented acetabular components. J Biomed Mater Res. 2001;55:433-441.
19. Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673-2682.
20. McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17-23.
21. Hercus B, Saeed S, Revell PA. Expression profile of T cell associated molecules in the interfacial tissue of aseptically loosened prosthetic joints. J Mater Sci Mater Med. 2002;13:1153-1156.
22. Baldwin L, Flanagan BF, Hunt JA. Flow cytometric measurement of phagocytosis reveals a role for C3b in metal particle uptake by phagocytes. J Biomed Mater Res A. 2005;73:80-85.
23. Martin SF, Merfort I, Thierse HJ. Interactions of chemicals and metal ions with proteins and role for immune responses. Mini Rev Med Chem. 2006;6:247-255.
24. Thakur SA, Hamilton R Jr, Pikkarainen T, Holian A. Differential binding of inorganic particles to MARCO. Toxicol Sci. 2009;107:238-246.
25. Rakshit DS, Lim JT, Ly K, et al. Involvement of complement receptor 3 (CR3) and scavenger receptor in macrophage responses to wear debris. J Orthop Res. 2006;24:2036-2044.
26. Tamaki Y, Takakubo Y, Goto K, et al. Increased expression of toll-like receptors in aseptic loose periprosthetic tissues and septic synovial membranes around total hip implants. J Rheumatol. 2009;36:598-608.
27. Beidelschies MA, Huang H, McMullen MR, et al. Stimulation of macrophage TNFalpha production by orthopaedic wear particles requires activation of the ERK1/2/Egr-1 and NF-kappaB pathways but is independent of p38 and JNK. J Cell Physiol. 2008;217:652-666.
28. Yamanaka Y, Abu-Amer Y, Faccio R, Clohisy JC. Map kinase c-JUN N-terminal kinase mediates PMMA induction of osteoclasts. J Orthop Res. 2006;24:1349-1357.
29. Caicedo MS, Desai R, McAllister K, et al. Soluble and particulate Co-Cr-Mo alloy implant metals activate the inflammasome danger signaling pathway in human macrophages: a novel mechanism for implant debris reactivity. J Orthop Res. 2009;27:847-854.
30. Dostert C, Pètrilli V, Van Bruggen R, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674-677.
31. Watanabe H, Gaide O, Pétrilli V, et al. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 2007;127:1956-1963.














