Managing Systemic Sclerosis and Its Complications
Systemic sclerosis (SSc), or scleroderma, is a chronic connective-tissue disease that has a multitude of clinical manifestations. These authors offer a guide to recognition and pathogenesis, as well as counsel about management.
ABSTRACT: Systemic sclerosis (SSc) has diverse clinical manifestations and mimics, complicating the diagnosis. Clinical presentation and outcomes are heterogeneous; in some patients, the disease follows a mild course, and in others, progressive disease develops. Skin tightening may be of greatest concern to patients, but internal organ dysfunction is common and may be life-threatening. Raynaud phenomenon is the initial symptom in most patients; the GI tract is the second most frequently involved organ. Together with pulmonary arterial hypertension, interstitial lung disease is the leading cause of death in SSc. Initial patient evaluation should include blood tests and screening for internal organ involvement. Many treatment strategies help physicians manage the complications and limit end-organ dysfunction. (J Musculoskel Med. 2011;28:380-387)
_______________________________________________________________________________________________
Systemic sclerosis (SSc), or scleroderma, is a chronic connective-tissue disease that has protean clinical manifestations. The cause is unknown, but it probably involves an environmental trigger in a genetically predisposed person. The pathophysiology of SSc involves vasculopathy, autoimmunity, and fibrosis that manifest as Raynaud phenomenon, autoantibody production, and varying degrees of skin thickening and internal organ dysfunction, respectively. The disease is most common in women in their fourth and fifth decades of life. The incidence of SSc in the US adult population is about 20 cases per million persons per year; the prevalence is estimated at 240 cases per million.1
Patients who have SSc may be classified in distinct clinical subsets that have characteristic patterns of skin and internal organ involvement and survival rates.2 Although to date there are no proven disease-modifying therapies for patients with SSc, many treatment strategies are highly effective for managing the disease complications and limiting end-organ damage.
In this article, we discuss the classification and diagnosis of SSc, the clinical manifestations-ranging from Raynaud phenomenon to skin, GI, lung, kidney, and heart involvement-the cause and pathogenesis, and evaluation. We also describe therapies for the disease and its complications.
TABLE 1



Scleroderma spectrum disorders
CLASSIFICATION AND DIAGNOSIS
The 2 main forms of SSc that affect adults are limited cutaneous (lcSSc) and diffuse cutaneous (dcSSc) (Table 1); classification is determined by the extent and pattern of skin involvement.3 If skin thickening involves areas distal to the elbows and knees, the patient has lcSSc; proximal skin fibrosis is characteristic of dcSSc. Facial involvement may be seen in either form.
However, many patients do not fit neatly into this dichotomous pattern. Up to 7 clinical subsets may be identified according to the presence of distinct serum autoantibodies; 4 subsets may be identified on the basis of gene expression patterns in skin.2,4
Although skin tightening often is of greatest concern to patients, internal organ dysfunction is common in the limited and diffuse forms and is potentially life-threatening. A small subset of patients have scleroderma sine scleroderma and lack skin tightening but experience characteristic internal organ dysfunction.
Localized forms of scleroderma affect mostly children and are not associated with significant internal organ complications (see Table 1). In addition, scleroderma mimics that are associated with skin induration but lack internal organ involvement need to be considered in establishing the diagnosis of SSc. Skin induration in the absence of Raynaud phenomenon, or sparing the fingers, is unlikely to be SSc.
Signs and symptoms related to gastroesophageal reflux disease (GERD) may bring patients with undiagnosed SSc to medical attention; initial symptoms may include chronic dry cough, hoarse voice, heartburn, pulmonary symptoms (eg, dyspnea on exertion or shortness of breath), arthralgias and diffusely swollen hands, and skin problems (eg, dry, itchy, hyperpigmented or hypopigmented skin). Misdiagnosis of undifferentiated polyarthropathy or carpal tunnel syndrome in patients who have early SSc is not uncommon.
TABLE 2



Systemic involvement in systemic sclerosis
CLINICAL MANIFESTATIONS
Raynaud phenomenon
The initial symptom in 70% of patients with SSc, Raynaud phenomenon develops in more than 95% of patients (Table 2). A diagnosis of Raynaud phenomenon is made when a patient provides a history of acral skin color changes precipitated by cold or emotional stress. In contrast to primary Raynaud syndrome, which typically presents during adolescence and does not lead to ischemic complications, secondary Raynaud phenomenon occurs later and often is complicated by ischemic tissue damage. Physical findings include reversible cyanosis and manifestations of ischemic damage, such as digital pitting, abnormal nail fold capillaroscopy, ischemic ulcers, and pterygium inversus unguis (Figure 1).
FIGURE 1

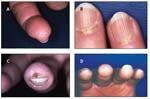
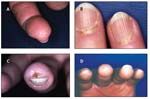
Dermatological manifestations of systemic sclerosis include digital pitting (A); visible punctate hemorrhages in nail fold capillaries (B); digital ulcer resulting from Raynaud phenomenon and chronic ischemia (C); and pterygium inversum unguis, a distal extension of the hyponychial (subungual) tissue that is anchored to the undersurface of the nail (D).
Skin involvement
Scleroderma, meaning hard skin, is the clinical hallmark of SSc. Skin changes usually begin on the fingers and hands; early swelling often is attributed to arthritis. Gradually, skin on the fingers (sclerodactyly), hands, and forearms becomes indurated. Over time, the process may stabilize (lcSSc) or continue to progress centripetally to include the up-per arms, chest, neck, abdomen, and trunk (dcSSc). Pigmentary changes are common and may be accompanied by intense pruritus and hair loss.
FIGURE 2



Sclerodactyly (tightened skin distal to the metacarpophlangeal joints) that causes flexion contractures of finger joints (A) and perioral furrowing on the face (B) are characteristic features of systemic sclerosis.
Physical findings include flexion contractures of the proximal interphalangeal joints; thickened, tight, and shiny skin on involved areas; and reduced oral aperture and deepening of perioral facial folds (radial furrowing) (Figure 2). Telangiectasias often occur on the face, buccal mucosa, chest, and hands.
GI involvement
The GI tract is the second most frequently involved organ in patients who have SSc; the entire tract from mouth to anus may be affected. Considerable pathology may be caused by esophageal and intestinal dysmotility, as well as GERD, even in asymptomatic patients.
Esophageal disease is especially common in patients who have the CREST (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) variant of lcSSc. Patients with CREST syndrome often have anticentromere antibodies and are at risk for pulmonary arterial hypertension (PAH). Heartburn and dysphagia are common; regurgitation/vomiting, sore throat, dental caries, and halitosis also may occur.
Because esophageal disease may be asymptomatic, acid suppression therapy should be started in all patients; in theory, such therapy may prevent stricture formation, aspiration pneumonitis, and Barrett esophagus. However, the precise role of acid suppression therapy in halting the progression of esophageal or pulmonary disease has not been established.5
Dysmotility of the intestinal tract may result in pseudo-obstruction and small-bowel bacterial overgrowth with vitamin B12 and folate deficiency, and malabsorption. Symptoms include bloating and abdominal pain, diarrhea/constipation, steatorrhea, and wasting.
Anemia may be a sign of vitamin deficiency resulting from bacterial overgrowth or chronic blood loss resulting from gastric antral vascular ectasia syndrome, or “watermelon stomach.” The latter term refers to the characteristic endoscopic appearance of longitudinal rows of sacculated and ectatic mucosal vessels in the antrum of the stomach; they resemble the stripes on a watermelon. Endoscopic laser coagulation and obliteration of vascular ectasia decrease the risk of rebleeding.
Chronic constipation is common and may result in intestinal impaction. A diagnosis of intestinal pseudo-obstruction often is made at the time of laparotomy, although nonsurgical treatments-including bowel rest, antibiotics, and judicious use of promotility agents-often are effective. Smooth muscle dysfunction in the anus causes fecal incontinence and is a common problem. Less common GI manifestations of SSc include asymptomatic pneumatosis cystoides intestinalis and wide-mouthed saccular diverticula of the colon.
Pulmonary manifestations
Interstitial lung disease (ILD) is common in SSc and together with PAH is the leading cause of death. African American men with SSc who have antibodies to topoisomerase (Scl-70), Th1/To, and fibrillarin are at increased risk for ILD, especially within 3 to 4 years after the diagnosis.2,6
The diagnosis of ILD often is made with screening high-resolution CT (HRCT) scans of the chest that demonstrate reticular opacification of the lung base or ground-glass opacification. More advanced ILD is associated with radiographic findings of honeycombing, traction bronchiectasis, and bilateral subpleural fibrosis most prominent in the lower lung zones. Unilateral or upper lobe abnormalities that are seen on HRCT scans may suggest infection or malignancy and may require evaluation by bronchoscopy, bronchoalveolar lavage (BAL) and, in some cases, open lung biopsy.
FIGURE 3



A lung biopsy specimen from a patient with systemic sclerosis demonstrates interstitial fibrosis with loss of normal lung architecture and pulmonary fibrosis (A). The pulmonary vasculature reveals thickening of vessel walls resulting from intimal hyperplasia, as well as intramural thrombosis. This lung biopsy specimen (B) demonstrates fibrosis that involves lung parenchyma with thickened interalveolar septae.
The pathological finding of nonspecific interstitial pneumonitis is most consistent with SSc-associated ILD (Figure 3). A restrictive pattern (reduced forced expiratory volume in 1 second, forced vital capacity [FVC], and total lung capacity) is most characteristic on screening pulmonary function tests (PFTs); FVC in the 50% to 75% of predicted range suggests moderate ventilatory restriction, and FVC less than 50% reflects severe disease. A proportionate reduction in carbon monoxide–diffusing capacity (DLCO) and FVC also may be observed; an FVC:DLCO ratio lower than 1:6 is consistent with ILD.5 The 10-year mortality rate for patients with severe restriction is 42%6; this high mortality underscores the need for early diagnosis and management of ILD.
Patients with both limited and diffuse forms of SSc are at risk for PAH, which may develop insidiously, in spite of having stable skin disease. Risk factors for severe PAH include limited cutaneous disease, older age, and elevated pulmonary artery pressures at initial evaluation.7 Postmortem examination reveals thickened pulmonary arteries with intimal thickening and luminal narrowing (see Figure 3).
Regular screening Doppler echocardiography and PFTs help detect PAH before the onset of cor pulmonale. The presence of PAH is suggested by pulmonary artery systolic pressure estimates higher than 35 mm Hg or DLCO lower than 55% of predicted in the absence of other causes. PAH characteristically causes a disproportionate reduction in DLCO, reflected by an FVC:DLCO ratio greater than 1.6.8
DLCO lower than 55% of predicted is 57% sensitive and 71% specific for PAH in patients with SSc in whom significant ILD has been excluded.9 The sensitivity of Doppler echocardiography for PAH approaches 90%; its specificity is 75%.8 Therefore, neither Doppler echocardiography nor DLCO measurements are by themselves sufficiently sensitive or specific to establish or rule out PAH. However, they are useful screening tools when performed serially as part of regular clinical assessments.
The only way to directly measure pulmonary artery pressures and evaluate response to treatment is right heart catheterization. PAH is present when mean pulmonary artery pressure, as measured at right heart catheterization, exceeds 25 mm Hg. Left heart catheterization may be performed concomitantly with right heart catheterization to rule out coronary artery disease and left ventricular diastolic dysfunction, which can cause pulmonary venous hypertension.
Renal disease
Before the introduction of angiotensin-converting enzyme (ACE) inhibitors, scleroderma renal crisis (SRC) was the most deadly complication of SSc, with an 85% 1-year mortality rate.10 Risk factors for SRC include older age, early disease and diffuse or rapidly progressing skin involvement, significant corticosteroid use, pregnancy, and the presence of speckled antinuclear antibody (ANA) and anti-RNA polymerase autoantibodies.10,11
SRC is characterized by a sudden onset of malignant hypertension that may be accompanied by chest pain with decompensated congestive heart failure (CHF) and papilledema. Roughly 10% of patients who have SSc with SRC have normotensive SRC, with blood pressures that are elevated from baseline but in the normotensive range.12
Laboratory studies show proteinuria, microangiopathic anemia, microscopic hematuria, and progressive oliguric renal failure. The clinical presentation of SRC may be difficult to distinguish from that of thrombotic thrombocytopenic purpura (TTP), but normal or slightly reduced levels of the metalloproteinase ADAMTS-13 suggest SRC, whereas TTP generally is associated with undetectable ADAMTS-13 levels.
The pathogenesis of SRC is attributed to endothelial cell injury that leads to intimal thickening in small renal arteries as well as platelet aggregation. Luminal narrowing decreases renal perfusion, which further activates the renin-angiotensin system. Malignant hypertension and more renal damage ensue.
Patients should be counseled to monitor their blood pressure at home because early recognition and treatment of SRC are critical. Prompt initiation and aggressive titration of a short-acting ACE inhibitor, such as captopril, may stabilize blood pressure and may prevent progressive renal damage. ACE inhibition with long-acting agents to improve adherence should be continued even in patients who require dialysis, because a significant number can discontinue dialysis for up to 2 years post-SRC.
Cardiac involvement
This is common in SSc but seldom a presenting manifestation. Cardiac Raynaud phenomenon may cause ischemia/reperfusion injury with attendant contraction band necrosis and myocardial fibrosis, leading to conduction abnormalities and arrhythmias and possibly left ventricular diastolic dysfunction.13 Ventricular diastolic dysfunction is an increasingly recognized cause of CHF in the aging US population, but CHF secondary to diastolic dysfunction from SSc-associated myocardial fibrosis is controversial.14,15
Pulmonary and renal complications of SSc are additional causes of CHF. Pericardial effusions are common but rarely lead to cardiac tamponade or hemothorax.
Musculoskeletal involvement
Concomitant polymyositis with weakness and elevations in aspartate aminotransferase, creatine kinase, and aldolase levels on laboratory studies develops in many patients, especially those with the diffuse form of the disease. Sclerodactyly and skin tightening may lead to joint contractures that impair mobility and predispose to traumatic ulcer formation, especially on the proximal interphalangeal joints. Tendon friction rubs that can be palpated over any joint, but often are palpated over the ankle, knee, hip, elbow, wrist, and metacarpophalangeal joints, are associated with diffuse disease and are a risk factor for SRC.
ETIOLOGY AND PATHOGENESIS
The cause of SSc is poorly understood. The disease involves a complex interplay of 3 seemingly distinct pathophysiological processes: small-vessel vasculopathy, autoimmunity, and fibrosis.16 Vasculopathy underlies Raynaud phenomenon as well as renal, pulmonary, and cardiac pathology. The pathogenesis of Raynaud phenomenon is multifactorial; it involves reversible vasospasm in small arteries and arterioles, which is followed by intrinsic vessel wall abnormalities with smooth muscle cell hypertrophy, intimal fibrosis, and endothelial cell apoptosis.17
TABLE 3



Systemic sclerosis clinical associations with scleroderma-specific autoantibodies
Autoimmunity in SSc is manifested by the presence of characteristic autoantibodies. Nearly all patients with SSc have a positive ANA, often with a nucleolar immunofluorescence pattern. Further testing for SSc-specific antibodies often reveals the presence of anticentromere and Scl-70 antibodies that are considered to be more specific for lcSSc and dcSSc, respectively (Table 3). The direct role of SSc autoantibodies in causing tissue damage is unclear because several studies have demonstrated autoantibody production in healthy family members and spouses of patients with SSc.18
Fibrosis of the skin and multiple internal organs is the pathological hallmark of SSc. Fibrosis is attributed to progressive accumulation of extracellular matrix proteins-primarily collagens-in affected tissues. Resident fibroblasts in lesional tissue, stimulated by transforming growth factor β, connective-tissue growth factor, platelet-derived growth factor, and related profibrotic cytokines and chemokines, become activated and transform into contractile myofibroblasts. Myofibroblasts synthesize collagen and other extracellular matrix proteins, which accumulate in affected organs, disrupting their function (see Figure 3).
EVALUATION
Initial evaluation of patients with SSc should include the following blood tests: serologies, including an ANA test and scleroderma-specific autoantibodies (anti-topoisomerase and anticentromere antibodies); a complete blood cell count to assess anemia; and a comprehensive chemistry panel to evaluate renal and liver function. The serum levels of brain natriuretic peptide may be elevated in patients who have PAH and is a useful screening test.19 Determining the erythrocyte sedimentation rate and C-reactive protein level also may be useful; elevation suggests vasculitis, malignancy, or overlap with another autoimmune disease.
To screen for the presence of internal organ involvement, we recommend that Doppler echocardiography, HRCT lung scanning, and full PFTs (including the 6-minute walk test) be performed on all patients at baseline. If echocardiography and PFT results are normal, serial annual studies are sufficient for disease monitoring. If there is concern about PAH and screening tests do not demonstrate abnormal pulmonary pressures, referral to a specialty center for right heart catheterization and hemodynamic evaluation is appropriate.
Screening for ILD is accomplished with routine PFTs and HRCT scanning. Findings of ventilatory restriction or radiographic evidence of ground-glass opacification, traction bronchiectasis, or honeycombing herald ILD. BAL may be indicated to rule out infection before immune suppression is started, but it is no longer used to establish a diagnosis or predict progression of SSc-ILD.20
MANAGEMENT
Severe heart, lung, renal, or GI tract involvement is most likely to develop within 3 years after the onset of dcSSc symptoms; therefore, early detection and prompt initiation of therapy are of greatest benefit.21 Lung involvement in lcSSc as well as SRC can occur even late in the disease course; thus, continued routine screening is essential.10
Currently, there is no proven disease-modifying agent to prevent or reverse fibrosis of the skin and internal organs. However, substantial progress has been made in the management of the organ-specific complications of SSc.
In particular, several agents provide relief for Raynaud phenomenon, GERD, and the sequelae of disordered intestinal motility. Most significantly, judicious early use of ACE inhibitors is highly effective in managing SRC; however, they should not be used to prevent SRC because evidence suggests that ACE inhibition before the onset of SRC may worsen outcomes.10
PAH-specific therapies (endothelin-1 receptor antagonists, phosphodiesterase-5 inhibitors, prostacyclin analogues) are approved only for use in patients with SSc who have undergone a right heart catheterization that has demonstrated a resting mean pulmonary artery pressure higher than 25 mm Hg. Their use in patients with ILD can increase ventilation-perfusion mismatch and may worsen symptoms. A recent retrospective study suggested that PAH-specific therapies may not be beneficial in this patient population.22
In patients with early ILD, use of oral and intravenous cyclophosphamide has been shown to induce a modest and transient improvement in FVC.23-25 In addition, dyspnea, skin thickening, and health-related measures of quality of life showed improvement.
Recently, a small study showed that autologous stem cell transplant improves skin disease, lung function, and health care related quality of life in carefully selected patients with SSc.26 The results of 2 larger stem cell clinical trials (Autologous Stem Cell Transplantation International Scleroderma and Scleroderma: Cyclophosphamide or Transplantation) of patients with SSc are forthcoming.
Lifestyle modification may greatly impact quality of life. In patients with Raynaud phenomenon, smoking cessation and avoidance of excess caffeine, amphetamine and dextroamphetamine, and cold exposure reduce the risk of digital ischemia. Head-of-the-bed elevation and avoidance of large, late meals can improve GERD symptoms. In addition, daily blood pressure monitoring can help detect SRC early so that ACE inhibition can be initiated promptly.
Corticosteroids have been shown to be associated with SRC. Therefore, if corticosteroids are to be used to manage an inflammatory arthritis or SSc/myositis overlap syndrome, they should be given at the lowest possible dose, and patients should be instructed to monitor their blood pressure.
Treatment of patients with SSc must be individualized to maximize benefit and reduce the risk of complications. The modest benefits of cyclophosphamide are coupled with the risk of hemorrhagic cystitis, bone marrow suppression, infection, infertility, and long-term malignancy. Stem cell transplant appears to be a promising treatment for some patients with SSc, but its use must be balanced against toxicity. The use of calcium channel blockers may exacerbate GERD symptoms, making angiotensin receptor II blockers, such as losartan, a better choice for the management of Raynaud phenomenon.
Proper management of SSc requires close monitoring and judicious use of organ-specific therapies. Referral to a center that can best provide integrated care by appropriate specialists often is beneficial and is strongly recommended. Clinical trials are under way to evaluate various immunomodulatory and antifibrotic approaches to treatment, including stem cell transplant.
Problems/comments about this article? Please send feedback.
References:
References
1. Mayes MD, Lacey JV Jr, Beebe-Dimmer J, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003;48:2246-2255.
2. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005;35:35-42.
3. LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202-205.
4. Milano A, Pendergrass SA, Sargent JL, et al. Molecular subsets in the gene expression signatures of scleroderma skin [published correction appears in PLoS One. 2008;3(10). doi:10.1371/annotation/05bed72c-c6f6-4685-a732-02c78e5f66c2]. PLoS One. 2008;3:e2696.
5. Ebert EC. Esophageal disease in scleroderma. J Clin Gastroenterol. 2006;40:769-775.
6. Steen VD, Conte C, Owens GR, Medsger TA Jr. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994;37:1283-1289.
7. Chang B, Schachna L, White B, et al. Natural history of mild-moderate pulmonary hypertension and the risk factors for severe pulmonary hypertension in scleroderma. J Rheumatol. 2006;33:269-274.
8. Steen V. Predictors of end stage lung disease in systemic sclerosis. Ann Rheum Dis. 2003;62:97-99.
9. Mukerjee D, St George D, Knight C, et al. Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology (Oxford). 2004;43:461-466.
10. Penn H, Howie AJ, Kingdon EJ, et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. QJM. 2007;100:485-494.
11. Steen VD. Scleroderma renal crisis. Rheum Dis Clin North Am. 2003;29:315-333.
12. Hudson M, Baron M, Lo E, et al. An international, web-based, prospective cohort study to determine whether the use of ACE inhibitors prior to the onset of scleroderma renal crisis is associated with worse outcomes-methodology and preliminary results. Int J Rheumatol. 2010;2010. pii:347402. Epub 2010 Sep 14.
13. Steen V. The heart in systemic sclerosis. Curr Rheumatol Rep. 2004;6:137-140.
14. Movahed MR, Ahmadi-Kashani M, Saito Y. Prevalence of suspected diastolic dysfunction in patients with a clinical diagnosis of congestive heart failure. Heart Fail Rev. 2005;10:263-264.
15. Aguglia G, Sgreccia A, Bernardo ML, et al. Left ventricular diastolic function in systemic sclerosis. J Rheumatol. 2001;28:1563-1567.
16. Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557-567.
17. Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol. 2005;17:752-760.
18. Mayes MD. Epidemiologic studies of environmental agents and systemic autoimmune diseases. Environ Health Perspect. 1999;107(suppl 5):743-748.
19. Nagaya N, Nishikimi T, Okano Y, et al. Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardiol. 1998;31:202-208.
20. Goh NS, Veeraraghavan S, Desai SR, et al. Bronchoalveolar lavage cellular profiles in patients with systemic sclerosis–associated interstitial lung disease are not predictive of disease progression. Arthritis Rheum. 2007;56:2005-2012.
21. Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000;43:2437-2444.
22. Le Pavec J, Girgis RE, Lechtzin N, et al. Systemic sclerosis–related pulmonary hypertension associated with interstitial lung disease: impact of pulmonary arterial hypertension therapies. Arthritis Rheum. 2011;63:2456-2464.
23. Tashkin DP, Elashoff R, Clements PJ, et al; Scleroderma Lung Study Research Group. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354:2655-2666.
24. Tashkin DP, Elashoff R, Clements PJ, et al; Scleroderma Lung Study Research Group. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007;176:1026-1034.
25. Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54:3962-3970.
26. Burt RK, Shah SJ, Dill K, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet. 2011;378:498-506.














