TKIs and the Resistance Challenge
Lung cancers with mutations in the epidermal growth factor receptor (EGFR) respond well to EGFR tyrosine kinase inhibitors (TKIs), but drug resistance invariably emerges, typically after a median of 10 to 14 months of treatment.
Several mechanisms of acquired resistance were discussed in a landmark paper by Zhou et al1, who noted that in more than 50% of cases, T790M is responsible for TKI resistance. T790M is a gatekeeper mutation that restores ATP binding and permits tyrosine kinase activity, even in the presence of inhibitors. Another major
mechanism of acquired resistance is compensatory amplification of the MET gene, which has been suggested to occur in more than 20% of cases of resistance.
Sequist et al2 elaborated on the subject of the evolution of drug resistance with a recent series of genetic and histological analyses of tumor biopsies from 37 patients with drug-resistant nonsmall cell lung cancer (NSCLC) carrying EGFR mutations. In addition to retaining all original activating EGFR mutations, some of the tumors
acquired the commonly occurring EGFR T790M mutation or MET gene amplification.2
Some tumors displayed unexpected genetic changes, however, including EGFR amplification, mutations in the PIK3CA gene, and a pronounced epithelial-to-mesenchymal transition. Five (14%) of the resistant cancers transformed from NSCLC to small cell lung cancer (SCLC).
Resistance was lost in three patients without the continued selective pressure of EGFR inhibitor treatment, meaning that responsiveness to a second round of EGFR inhibitor treatment was restored.2
One surprising finding from research on the molecular mechanisms of resistance is that while some mutations in the EGFR gene are activating and drive tumor formation, others could actually be governing intrinsic resistance against EGFR inhibitors. Intrinsic resistance is also reportedly conferred by the presence of mutations in the KRAS gene
Other theories posited as explanations for acquired resistance to EGFR inhibitors include ubiquitination, which targets EGFR for destruction in the cell; the epithelial-to-mesenchymal transition, in recognition of the fact that mesenchymal cells are more resistant; oncogenic shift, involving increased expression of alternative ErbB receptors; and activation of alternative signaling pathways, including the Akt/mTOR cascade. As a result, EGFR inhibitors are also being tested in combination with
other agents, including vascular endothelial growth factor receptor and MET TKIs.

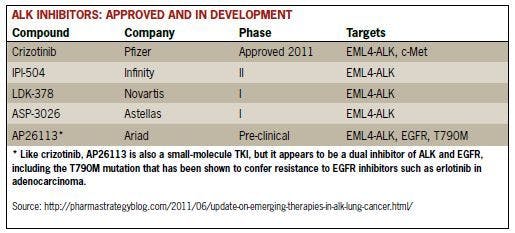
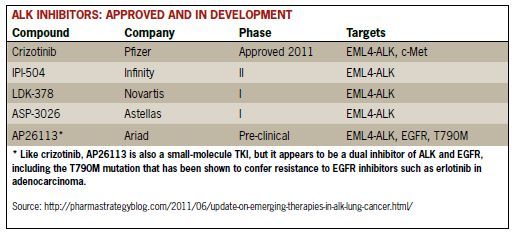
Acquired Resistance to Crizotinib
Acquired resistance to crizotinib has been reported in patients with ALK gene-rearranged NSCLC who had previously shown robust responses to the drug. In a study to gain insight into the mechanisms of intrinsic and acquired crizotinib resistance in ALK-positive NSCLC patients who showed radiologic progression while on crizotinib,
Doebele et al3 identified three responsible processes: somatic domain mutations, ALK
gene fusion copy number gain (CNG), and the emergence of separate and new oncogenic drivers. Based upon molecular analyses of tissue from 11 patients, the investigators found the following:
• Four patients (36%) developed secondary mutations in the tyrosine kinase domain of ALK. In two of the four patients, the cause was a novel mutation in the ALK domain.
• Two patients (one with a resistance mutation), had evidence of new-onset ALK CNG.
• One patient had evidence of outgrowth of EGFR-mutant NSCLC without evidence of an ongoing ALK gene rearrangement.
• Two patients each had a KRAS mutation, one of which appeared to be lacking evidence of an ALK gene rearrangement.
• In one patient’s tumor, there was evidence of the emergence of an ALK gene fusion-negative tumor, compared with baseline, but without an identifiable alternative driver.
• Two patients retained ALK-positive status, but with no identifiable resistance mechanism.
Writing in his blog (cancergrace.org) about these findings by the researchers from the University of Colorado, H. Jack West, MD, suggests two potential mechanisms for acquired resistance appear to be operating. The first is development of a second cancer driver coexisting with the original one in the cancer cell. “Under evolutionary pressure of a treatment with an EGFR tyrosine kinase inhibitor (TKI), …the cancer cells still have the driver mutation (EGFR, in this example) but develop a competing and overriding feature that confers resistance.”4
In the second mechanism scenario, “selection pressure” of effective treatment may lead to a new oncogenic driver mutation. “The idea here is that there are different subsets of cancer cells.” And, prior to treatment with a targeted therapy “… the cancer may be comprised of primarily ALK rearrangement positive cancer cells, while a small minority has a KRAS mutation.” After ALK inhibitor therapy, ALK-positive cells die, yielding a cancer that is now growing and comprised predominantly
of KRAS mutation positive cancer.4
There is significant clinical relevance to this work, West says, even if the implications remain unproven at this time. “We’ve seen examples of situations in which the cancer, or at least some areas of the cancer, have morphed into a new version, sometimesa different subtype of lung cancer entirely. Even looking at the small number of cases here, we can see a complex array of possibilities. Our knowledge of what’s possible is growing on a case by case basis, but it is helping direct us to literally individualized recommendations for patterns that are becoming cancer and patient-specific.”4
West points to the potentially significant value to doing repeat biopsies, since these results may help shape subsequent treatment decisions.
References
1. Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070-1074.
2. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26.
3. Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472-1482.
4. West J. New Insights on mechanisms of resistance to XALKORI (crizotinib): implications for molecular oncology. January 13, 2012. Available at http://cancergrace.org/
lung/2012/01/13/mechanisms-of-criz-resistance/ Accessed March 29, 2012.









 May Offer Over PPIs, with Adelina Hung, MD")





