Article
Diagnosis and management of gout: An update
Gout is the most common inflammatory arthritis. Arthrocentesis and identification of negatively birefringent monosodium urate crystals from aspirate is the gold standard for diagnosis.
Gout, “the king of diseases and the disease of kings,” was one of the earliest disorders to be recognized as a clinical entity.1 No longer discriminating among socioeconomic classes, it has more than doubled in prevalence over the past 2 decades.2 A disorder of purine metabolism, gout now is the most common inflammatory arthropathy. It occurs in men more than in women (ratio, about 3 or 4:1).2
Even in this setting of a rising prevalence, primary care management of gout does not always adhere to evidence-based best practice.3 In addition, a significant number of patients do not respond to appropriate therapy or do not achieve optimal response to treatment, even in the hands of experienced rheumatologists (R. T. Keenan, M. H. Pillinger, unpublished data).
Recently, after almost 50 years of dormancy, new therapeutic agents for the management of gout have entered the market or are in clinical development. In this article, we review current guidelines for the diagnosis and management of gout.
DIAGNOSIS
The diagnosis of gout is not always easy to make. The original American College of Rheumatology criteria4 have been shown to have limited validity (sensitivity, 0.80; specificity, 0.64; positive predictive value, 0.80; and negative predictive value, 0.65).5 More recently, the European League Against Rheumatism (EULAR) also developed recommendations.6
Visualization of crystals
The gold standard for making a diagnosis of gouty arthropathy is visualization of negatively birefringent, needle-shaped crystals. This is done with arthrocentesis and analysis of synovial fluid (SF) or tophus aspirate and identification of monosodium urate (MSU) crystals, particularly intracellular crystals in neutrophils.6,7 MSU crystals also may be identified in SF obtained from asymptomatic joints, especially the knees and the first metatarsophalangeal (MTP) joints.7,8
Of note, acute gouty arthritis may coexist with another joint disease, such as septic arthritis or pseudogout. Therefore, arthrocentesis should be performed in almost all circumstances.
Serum uric acid (SUA) levels may be obtained during an acute attack (a high SUA level supports the possibility that a patient has gout). However, SUA levels may be paradoxically low during an acute attack (because of an increase in renal excretion); they typically are highest about 2 weeks after.
Previously proposed criteria
A thorough history helps distinguish an acute gout attack from other causes of acute arthritis. Previously proposed clinical, radiographic, and laboratory criteria include the following:
•Identification of MSU crystals from the SF of the affected joint.
•Negative Gram stain and culture results from the SF.
•A history of 1 or more episodes of monarticular arthritis, followed by intercritical periods free of symptoms.
•Maximum inflammation within 12 to 24 hours after the onset of the attack.
•Rapid resolution of synovitis after colchicine or NSAID therapy.
•Unilateral first MTP joint attack (podagra), especially if it is the first event.
•Hyperuricemia.
•Subcortical bone cysts on plain radiographs.6
Natural history
The natural history of gout may be translated into 3 clinical periods. They are: (1) asymptomatic hyperuricemia; (2) episodes of acute attacks of gout, with asymptomatic intervals (intercritical gout); and (3) chronic gouty arthritis.
Chronic hyperuricemia. This is the most important risk factor for gout.6 The risk of acute gout rises with the SUA concentration. In patients with SUA concentrations of 6.1 mg/dL (540 µmol/L) or higher, the cumulative incidence of gouty arthritis is 22% after 5 years.6,9 However, many persons with high SUA levels do not ever have gout, indicating that factors other than hyperuricemia play a role in the development of clinical symptoms.
Acute gouty arthritis. Episodes of acute gouty arthritis often begin at night or in the early morning; pain and swelling escalate rapidly. The joint quickly becomes warm, red, and tender (“calor, rubor, dolor, et tumor”), potentially mimicking cellulitis or even a septic joint.
Acute attacks typically affect the first MTP joint (up to 50% of first-time attacks), but tarsal joints, ankles, knees, elbows, and interphalangeal (IP) joints all are frequently affected. Rarely, the initial attack is polyarticular
(3% to 14% of cases); acute attacks infrequently affect the shoulders or hips.
In most cases, unmanaged gout resolves within a few days. Most patients have a second attack within 6 months to 2 years, although only a single episode occurs in some. Subsequent attacks often last longer than the initial attack, and they are more likely to affect proximal joints, including the knees and carpal and IP joints.10
Chronic gouty arthritis. When left unmanaged, acute attacks of gout can lead to chronic gout. This condition is characterized by chronic destructive polyarticular involvement with low-grade joint inflammation, joint deformity, and tophi-MSU crystals surrounded by chronic mononuclear and giant-cell reactions.11 Tophaceous gout develops within 5 years of the onset of gout in 30% of untreated patients.11
Tophi often are seen in the helices of the ears, over the olecranon processes, on the Achilles tendons, within and around the toe or finger joints, around the knees, and within the prepatellar bursae. The skin overlying the tophus may ulcerate and extrude white, chalky material composed of MSU crystals. Tophi are painless and rarely become infected.
Imaging

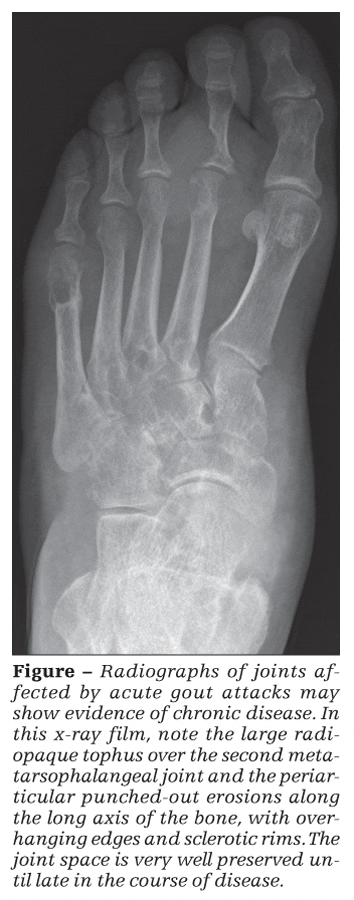
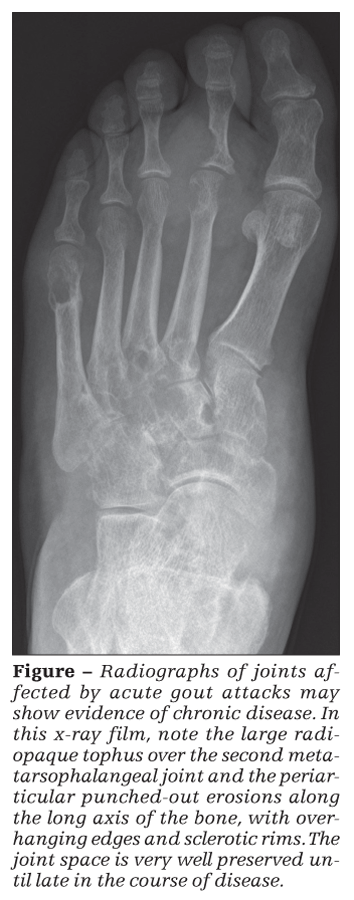
Radiographs of joints affected by acute gout attacks typically are not useful for the diagnosis of acute gout. However, they may show evidence of chronic disease (Figure).
Musculoskeletal ultrasonography (MSUS) continues to gain popularity in the evaluation of various musculoskeletal diseases, including crystal-induced arthropathy. MSUS has the capacity to visualize intra-articular crystal deposits with a characteristic hyperechoic enhancement of the outer surface of the hyaline cartilage (the “double contour sign”). Although MSUS may be useful in the diagnosis of gout, it has limitations, paramount of which is the inability to differentiate between the type of crystal deposition and the presence or absence of infection.
CT and MRI may be useful in confirming the presence of erosion and tophi.12 Since the use of both imaging modalities has increased, the prevalence of axial skeleton gout also has increased.13
TREATMENTAcute gout
Phagocytosis of MSU crystals is the inciting factor of acute gouty arthritis. The urate crystals are recognized as foreign and taken up by macrophages, inciting an inflammatory response (activation of the NLRP3 inflammasome). This activation triggers a cascade that results in the release of interleukin (IL)-1 and other inflammatory cytokines.14 The release of the cytokines rapidly ignites a broader inflammatory response and the infamous redness, pain, and swelling of an acute gout flare.

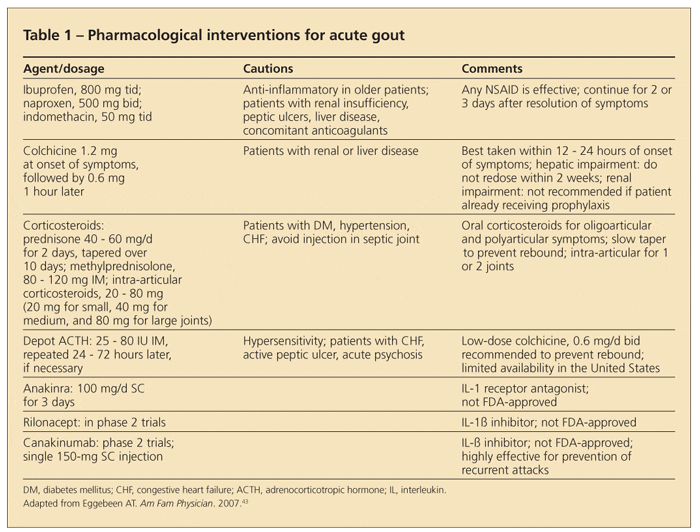
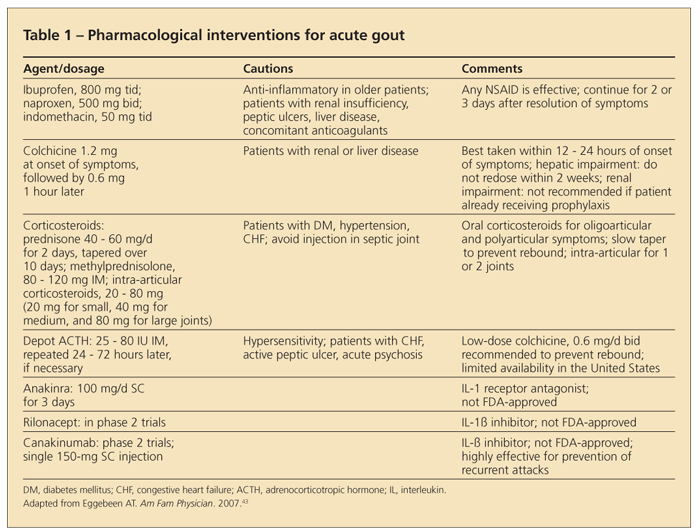
The goal of therapy for acute gout is to stop this inflammatory process. Therefore, the faster the attack is recognized and medications (including NSAIDs, colchicine, corticosteroids, and even IL-1 inhibitors) are administered, the quicker and easier the attack is to abate (Table 1).
NSAIDs. For most patients, NSAIDs are the preferred agent for managing acute gout. All NSAIDs appear to be equally effective when used at a full anti-inflammatory dose. The most frequently used regimens are ibuprofen, 800 mg tid; naproxen, 500 mg bid; and indomethacin, 50 mg tid.15-18
Having adequate dosing and duration is important. Treatment should be continued until the flare has resolved and then reduced in tapered doses for at least 2 or 3 days after all overt signs of inflammation are gone. NSAIDs have the advantage of acting as an analgesic as well as an anti-inflammatory, but they may be somewhat contraindicated in patients who have hypertension, congestive heart failure (CHF), renal insufficiency, gastritis/ulcers, and a number of other conditions.
Colchicine. To maximize response time while minimizing adverse effects, some clinicians managing acute gout add an intermediate dosing regimen of colchicine (0.6 mg every 6 to 8 hours) to their patients' NSAID regimens. The use of a proton pump inhibitor can improve GI tolerance of NSAIDs and reduce the likelihood of gastric bleeding, but it may not avoid other concerns.
Although colchicine has been used for gout management since the 6th century ad,19 only 1 year has passed since its FDA approval for acute and chronic gout. As a result, dosing guidelines and the evidence basis for colchicine in acute gout management have advanced.
Historically, colchicine has been given orally or intravenously. Oral colchicine is taken every 1 to 2 hours for acute gout flares until toxicity occurs. When the first signs and symptoms associated with toxicity occur-including diarrhea, nausea, and vomiting-dosing is stopped. Intravenous dosing bypasses the GI tract and therefore is associated with less GI intolerance but at the risk of severe cytopenia, acute renal failure, and disseminated intravascular coagulopathy. The high-dose oral regimen has fallen out of favor; intravenous colchicine was taken off the market in 2008.
The low-dose colchicine regimen has been found to be as effective as the high-dose colchicine regimen and much less toxic. In a large, randomized, controlled, multicenter trial that compared low-dose and extended-dose colchicine regimens, the results strongly supported the use of 1.2 mg within 12 hours of the onset of the attack, followed by 0.6 mg 1 hour later.20
Corticosteroids. These potent anti-inflammatory agents can be highly effective at abrogating acute gout. Intra-articular corticosteroids may be particularly useful in managing acute gout in a single joint or bursa and in cases in which the systemic corticosteroid load needs to be minimized. Care must be taken to rule out infection before injecting corticosteroids into the joint; this may mean performing joint aspiration and injection separately, an approach that patients do not always appreciate. Both oral and intravenous corticosteroids are very effective, especially in patients who are experiencing polyarticular attacks or have contraindications to colchicine and NSAIDs.
Most rheumatologists prescribe about 40 mg/d of prednisone with a slow taper (similar to management of an acute asthma attack) to avoid rebound attacks after corticosteroid withdrawal. Although treatment periods with oral corticosteroids typically are fairly brief (less than a week), potential contraindications-such as diabetes mellitus, hypertension, and heart failure-still must be considered.
Adrenocorticotropic hormone (ACTH). A single intramuscular injection of depot ACTH gel (25 to 80 IU) repeated 24 to 72 hours later, if needed, also is a very powerful anti-inflammatory, and it may be more effective than oral corticosteroids. ACTH stimulates the adrenal cortex to produce corticosteroids, and it may suppress the acute inflammatory response by activating melanocortin receptor 3.21 However, access to ACTH gel in the United States currently is limited.
New therapies. Acute gout therapy has not changed for decades, but there are medications on the horizon that may come to fruition. Development of potential new therapies has capitalized on evolving understanding of the pathophysiology of the inflammatory response in acute gout.
Targeting the secretory product of the inflammasome, a pilot study of 10 patients with chronic refractory gouty inflammation given the soluble IL-1 receptor antagonist anakinra (100 mg/d SC for 3 days) suggested good overall responses, although results of larger randomized, controlled trials are needed.22 Rilonacept, a soluble IL-1 receptor-Fc fusion protein,23 and canakinumab, a fully human monoclonal anti–IL-1β antibody,24 have shown promising results both as treatment for patients with acute, difficult-to-manage gout attacks and prevention of recurring attacks.
Chronic gout
The goals of chronic gout treatment are to eliminate the recurrence of acute attacks and to decrease or eliminate the tophus burden in the joints and soft tissues. These goals are achieved by lowering the SUA level to less than 6 mg/dL.25
The first step in managing gout is patient education. SUA levels may be lowered modestly (by 1 or 2 mg/dL) with lifestyle changes, such as weight loss, avoidance of purine-rich foods (organ meats, shellfish, beer), avoidance of processed foods that have high-fructose sweeteners, elimination or reduction of alcohol consumption (beer and liquor), and even increased intake of vitamin C.26
Medications also may play a role in gout management by raising or lowering SUA concentrations with stimulation or inhibition of the urate transporter URAT1 in the proximal tubule of the kidney, respectively.27 The use of diuretic drugs (eg, thiazides and loop diuretics) increases the risk of gout attacks, and the occurrence of arthritis shortly after the start of SUA-lowering therapy is well established.28 In contrast, some medications (eg, losartan) inhibit URAT1 and subsequently lower SUA levels.27 Although lifestyle interventions and daily low-dose colchicine or NSAID prophylaxis may be all that is needed in some patients with early mild gout, such interventions typically do not replace the need for SUA-lowering drug therapy to prevent frequent attacks (more than 2 per year).

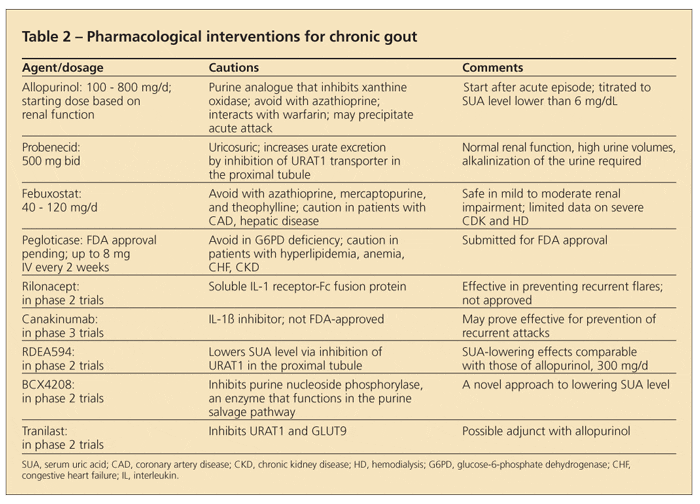
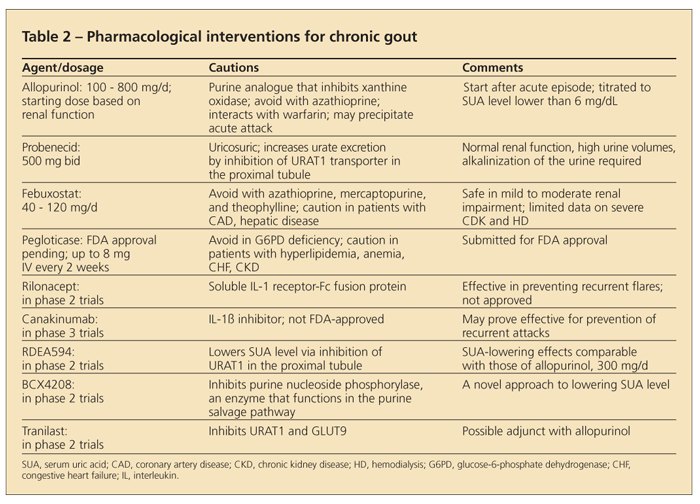
SUA-lowering agents frequently used to manage chronic gout include purine and nonpurine inhibitors of xanthine oxidase (allopurinol, febuxostat) and uricosuric agents (probenecid, sulfinpyrazone, benzbromarone) (Table 2). Also, new medications on the horizon may be used instead of or in addition to those already approved for use in chronic gout.
Allopurinol, the most frequently used SUA-lowering agent in the treatment of patients with gout, is FDA-approved for doses up to 800 mg/d. In clinical practice, nonadherence with allopurinol treatment was elucidated to be a problem in about 50% of patients in the first year of therapy.29
Allopurinol is widely underdosed in clinical practice-the vast majority of allopurinol prescriptions are for 300 mg/d,30 even though higher doses often are required to achieve an SUA target level (lower than 6 mg/dL). Such dosing practices stem from limited 30-year-old data about potentially serious adverse effects in patients with chronic kidney disease (CKD).31 The adverse effects, which include rash, cytopenia, and fever, are uncommon.
Although most serious adverse effects of CKD occur within the first 6 to 8 weeks of allopurinol use, long-term monitoring is required. Daily doses ranging from 600 to 1000 mg can be used safely in patients with CKD.
The EULAR currently recommends dose adjustment in patients with CKD.6,31 A good rule to follow is to start with a lower dose, then titrate to the dose until the target SUA level is achieved, all the while monitoring the patient for signs and symptoms of hypersensitivity.
Interactions with other medications can occur. For example, azathioprine is metabolized by xanthine oxidase; concomitant use of the drugs can raise azathioprine levels and result in bone marrow toxicity. Consequently, use of the drugs together is discouraged.
Febuxostat is a potent, nonpurine, selective inhibitor of xanthine oxidase.32 More significantly, it is primarily metabolized by the liver. Dosing is limited to 40 or 80 mg/d, making dose adjustment unnecessary in patients with mild to moderate kidney impairment.33 Febuxostat also does not affect pyrimidine metabolism, and in contrast to allopurinol, it does not have the potential to contribute to certain drug toxicities. The results of clinical trials unequivocally established the superiority of febuxostat over 300 mg/d of allopurinol in achieving an SUA target level of lower than 6 mg/dL and reducing tophaceous deposits in most of the patient population that was studied.
Uricase therapy:
a “biologic” option
Humans, primates, New World monkeys, and Dalmatians lack uricase (the enzyme that converts uric acid to the more soluble and excretable allantoic acid).34 It was not until about 50 years ago that the use of exogenous uricase in humans would be realized.35
Limited reports and pilot studies have evaluated off-label use of the non-PEGylated recombinant fungal enzyme rasburicase36,37-which is FDA-approved for single-course therapy in tumor lysis syndrome-in severe, chronic gout. However, rasburicase is not practical for long-term use because it is highly antigenic and has a short half-life.36 Also, at about $8000 per dose, the cost of rasburicase is prohibitive.
A recent advance has been made with a recombinant porcine-baboon uricase appended (PEGylated) to the protein chains of methoxypolyethylene glycol. The resultant pegloticase has been found to rapidly decrease SUA levels and debulk tophi in weeks to months (rather than the months to years seen with xanthine oxidase inhibitors taken at conventional doses), subsequently restoring lost articular function in many patients.38
Frequent early acute gout flares occurred (in up to about 80% of patients) in the first few months of pegloticase therapy but tapered off with more prolonged therapy in responders.38 Infusion reactions were moderate to severe in about 8% to 11% of patients; they included flushing, urticaria, and hypotension and, by undefined mechanisms, noncardiac chest pain and muscle cramping.
It still is not fully clear whether CHF, anemia, hyperlipidemia, CKD, and other comorbidities influence uricase. As an “induction therapy,” uricase ultimately could be replaced by less intensive maintenance oral SUA-lowering therapy with other agents, once resolution of clinically detectable tophi and gross synovitis is achieved. Pegloticase is awaiting FDA approval, and although there may be long-term safety issues, its potential for rapidly eliminating the tophus burden and reducing morbidity is indisputable.
With advances in understanding of the mechanism of urate metabolism and excretion, several potential medications currently are in phase 2 clinical trials (Table 2).39-42 RDEA594 and tranilast lower SUA levels via inhibition of URAT1 in the proximal tubule of the kidney.40,41 Tranilast is being studied as an adjunct to allopurinol and RDEA594 as monotherapy.41 BCX4208 exploits purine metabolism by inhibiting purine nucleoside phosphorylase.42
References:
References
1. Choi H. Epidemiology of crystal arthropathy. Rheum Dis Clin North Am. 2006;32:255-273, v.
2. Lawrence RC, Felson DT, Helmick CG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States, part II. Arthritis Rheum. 2008;58:26-35.
3. Pal B, Foxall M, Dysart T, et al. How is gout managed in primary care? A review of current practice and proposed guidelines. Clin Rheumatol. 2000;19:21-25.
4. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895-900.
5. Janssens HJ, Janssen M, van de Lisdonk EH, et al. Limited validity of the American College of Rheumatology criteria for classifying patients with gout in primary care. Ann Rheum Dis. 2010;69:1255-1256.
6. Zhang W, Doherty M, Pascual E, et al. EULAR evidence based recommendations for gout, part I: diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1301-1311.
7. Pascual E, Doherty M. Aspiration of normal or asymptomatic pathological joints for diagnosis and research: indications, technique and success rate. Ann Rheum Dis. 2009;68:3-7.
8. Pascual E. Persistence of monosodium urate crystals and low-grade inflammation in the synovial fluid of patients with untreated gout. Arthritis Rheum. 1991;34:141-145.
9. Saag KG, Choi H. Epidemiology, risk factors, and lifestyle modifications for gout. Arthritis Res Ther. 2006;(8 suppl 1):S2.
10. Liebman SE, Taylor JG, Bushinsky DA. Uric acid nephrolithiasis. Curr Rheumatol Rep. 2007;9:251-257.
11. Schumacher HR Jr, Becker MA, Palo WA, et al. Tophaceous gout: quantitative evaluation by direct physical measurement. J Rheumatol. 2005;32:2368-2372.
12. Dalbeth N, Clark B, Gregory K, et al. Computed tomography measurement of tophus volume: comparison with physical measurement. Arthritis Rheum. 2007;57:461-465.
13. Konatalapalli RM, Demarco PJ, Jelinek JS, et al. Gout in the axial skeleton. J Rheumatol. 2009;36:609-613.
14. Martinon F, Petrilli V, Mayor A, et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237-241.
15. Janssens HJ, Janssen M, van de Lisdonk EH, et al. Use of oral prednisolone or naproxen for the treatment of gout arthritis: a double-blind, randomised equivalence trial. Lancet. 2008;371:1854-1860.
16. Man CY, Cheung IT, Cameron PA, Rainer TH. Comparison of oral prednisolone/paracetamol and oral indomethacin/paracetamol combination therapy in the treatment of acute goutlike arthritis: a double-blind, randomized, controlled trial. Ann Emerg Med. 2007;49:670-677.
17. Terkeltaub RA. Colchicine update: 2008. Semin Arthritis Rheum. 2009;38:411-419.
18. Schumacher HR Jr, Boice JA, Daikh DI, et al. Randomised double blind trial of etoricoxib and indometacin in treatment of acute gouty arthritis. BMJ. 2002;324:1488-1492.
19. Nuki G, Simkin PA. A concise history of gout and hyperuricemia and their treatment. Arthritis Res Ther. 2006;(8 suppl 1):S1.
20. Terkeltaub RA, Furst DE, Bennett K, et al. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62:1060-1068.
21. Getting SJ, Lam CW, Chen AS, et al. Melanocortin 3 receptors control crystal-induced inflammation. FASEB J. 2006;20:2234-2241.
22. So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28.
23. Terkeltaub R, Sundy JS, Schumacher HR, et al. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis. 2009;68:1613-1617.
24. So A, De Meulemeester M, Pikhlak A, et al. Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis. Arthritis Rheum. 2010 Jun 8; [Epub ahead of print].
25. Schumacher HR Jr, Chen LX. Newer therapeutic approaches: gout. Rheum Dis Clin North Am. 2006;32:235-244, xii.
26. Stein HB, Hasan A, Fox IH. Ascorbic acid-induced uricosuria: a consequency of megavitamin therapy. Ann Intern Med. 1976;84:385-388.
27. Choi HK, Mount DB, Reginato AM. Pathogenesis of gout. Ann Intern Med. 2005;143:499-516.
28. Hunter DJ, York M, Chaisson CE, et al. Recent diuretic use and the risk of recurrent gout attacks: the online case-crossover gout study [published correction appears in J Rheumatol. 2006;33:1714]. J Rheumatol. 2006;33:1341-1345.
29. Riedel AA, Nelson M, Joseph-Ridge N, et al. Compliance with allopurinol therapy among managed care enrollees with gout: a retrospective analysis of administrative claims. J Rheumatol. 2004;31:1575-1581.
30. Sarawate CA, Brewer KK, Yang W, et al. Gout medication treatment patterns and adherence to standards of care from a managed care perspective. Mayo Clin Proc. 2006;81:925-934.
31. Chao J, Terkeltaub R. A critical reappraisal of allopurinol dosing, safety, and efficacy for hyperuricemia in gout. Curr Rheumatol Rep. 2009;11:135-140.
32. Okamoto K, Eger BT, Nishino T, et al. An extremely potent inhibitor of xanthine oxidoreductase: crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem. 2003;278:1848-1855.
33. Becker MA, Schumacher HR, Espinoza LR, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res Ther. 2010;12:R63.
34. Watanabe S, Kang DH, Feng L, et al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension. 2002;40:355-360.
35. Kissel P, Lamarche M, Royer R. Modification of uricaemia and the excretion of uric acid nitrogen by an enzyme of fungal origin. Nature. 1968;217:72-74.
36. Terkeltaub R. Learning how and when to employ uricase as bridge therapy in refractory gout. J Rheumatol. 2007;34:1955-1958.
37. Richette P, Briére C, Hoenen-Clavert V, et al. Rasburicase for tophaceous gout not treatable with allopurinol: an exploratory study. J Rheumatol. 2007;34:2093-2098.
38. Sundy JS, Becker MA, Baraf HS, et al. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum. 2008;58:2882-2891.
39. Study Utilizing Rilonacept in Gout Exacerbations (SURGE). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT00855920. Accessed September 9, 2010.
40. RDEA594, a novel uricosuric agent, significantly reduced serum urate levels and was well tolerated in a phase 2a pilot study in hyperuricemic gout patients. Rheumatology Aco. http://acr.confex.com/acr/2009/webprogram/Paper15506.html. Accessed September 9, 2010.
41. Study of tranilast alone or in combination with allopurinol in subjects with hyperuricemia. ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01052987?term=tranilast&rank=2. Accessed September 9, 2010.
42. Study to evaluate the urate-lowering activity and safety of oral BCX4208 administered in subjects with gout. ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT00985127?term=BCX4208&rank=2. Accessed September 9, 2010.
43. Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.












