How Should I Manage This Man's Red, Swollen Toe?
Examining the right foot of a 54-year-old male reveals a red, swollen, warm, and exquisitely tender right first toe joint. What is the differential diagnosis?

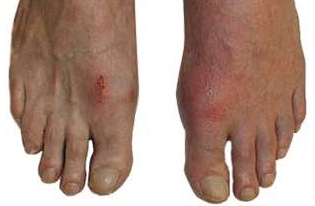
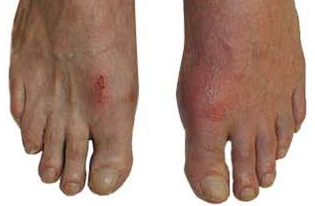
A 54-year-old male patient is seen for a 2-day history of severe right first toe pain. He denies a past medical history of fever, chills, or trauma, and his history is only notable for hypertension and impaired fasting glucose. His current medications are once-daily 25 mg hydrocholorothiazide and once-daily 81 mg enteric-coated aspirin. Examining the right foot reveals a red, swollen, warm, and exquisitely tender right first toe joint.
What is the differential diagnosis?
The patient has monoarthritis affecting the big toe joint, which is technically podagra — a term that already implies gout is the cause. The reason is that within this clinical setting of an older male with hypertension and hyperglycemia who is taking hydrochlorothiazide and aspirin — 2 agents that may interfere with uric acid excretion — gout is overwhelmingly the likely diagnosis. The other confirming diagnostic features are the rapid onset, involvement of the first metatarsophalangeal joint (MTP), and redness and swelling. In at least 80% of patients, the first gout attack involves only a single joint, and the first MTP is the affected joint more than 50% of the time.
Nevertheless, it's still important to consider septic arthritis, since the symptoms and findings can be indistinguishable from gout. If the patient had a history of recent infection or sepsis — including fever, chills, and elevated white blood cell (WBC) count preceding the attack — then I would definitely aspirate the joint and send the fluid for laboratory analysis, Gram stain, and culture.
Pseudogout, or crystal-induced synovitis caused by calcium pyrophosphate dehydrate, is a rare cause of first MTP inflammation, in which case the wrist and knee are the joints more likely to be affected. In a young woman, gout is very unlikely, so a more likely cause would be basic calcium phosphate or calcific periarthritis, either in the joint or the surrounding tissue. This can be diagnosed by the presence of stippled calcification on X-rays.
What diagnostic studies would you recommend?
If the capacity for joint aspiration and examination of synovial fluid under compensated polarizing microscopy is readily available, then joint fluid analysis is highly recommended, as it remains the gold standard for diagnosis. Gout crystals are seen in more than 85% of aspirates, and the specificity for the diagnosis of gout is 100%.
If joint fluid analysis is not readily available, then the clinician can rely on a clinical model developed in the Netherlands, where predefined variables are assigned scoring values:
- Male sex (2 points)
- Previous patient-reported gout attacks (2 points)
- Onset within 1 day (0.5 points)
- Joint redness (1 points)
- First MTP involvement (2.5 points)
- Hypertension or cardiovascular disease features (1.5 points)
- Serum uric acid >5.88 mg/dL (3.5 points)
In the Netherlands study, patients who scored low (≤4 points), intermediate (>4 to <8 points), and high (≥8 points) had a gout probability of 2.2%, 31.2%, and 82.5%, respectively. Most clinical models without joint aspiration have been shown to be approximately 70% sensitive and 85% specific.
I would typically order tests for complete blood count (CBC), comprehensive metabolic panel, uric acid, erythrocyte sedimentation rate (ESR), and C-reactive protein (CRP) in this setting.
The patient’s CBC reveals hemoglobin 15.1 g/d, hematocrit 46.8, and WBC count 7.74 with normal differential. His Westergren ESR is 30 mm/hr (NL 0 to 22 mm/hr) and CRP 1.0 mg/dL (N <0.8). His chemistries include a fasting glucose of 115, serum creatinine 1.1 mg/dL, BUN 22 mg/dL ALT 30 U/L, ALT 36 U/L, and alkaline phosphatase 88 U/L, while his uric acid is 6 mg/dL. Is joint aspiration necessary?
The laboratory studies are characteristic of an inflammatory episode, but serum uric acid is 6 mg/dL. A serum uric acid under 7 mg/dL — where the solubility of uric acid crystals occurs at 37 degrees Celsius — is seen in 50% of patients at the time of an acute gout attack. I recommend getting a second serum urate measurement 2 weeks after the initial attack if the first one is under 7 mg/dL.
If joint aspiration is not performed, then it is important to ensure that the attack ends within 72 hours. If there are residual redness and inflammation, then joint aspiration for sepsis becomes mandatory.
How would you initially manage the patient?
Acute gout is one of life’s most painful experiences. It has even been likened to a red hot poker getting applied to the toe. Therefore, the prompt yet safe relief of pain and swelling is the immediate goal. I start with a rapid-acting nonsteroidal anti-inflammatory drug (NSAID) like indomethacin, which I typically prescribe in an initial dose of 100 mg followed by 50 mg administered 8 hours later. After those doses, I keep the patient on 50 mg 3 times a day (tid) for the next 2 to 3 days. When relief becomes apparent, I reduce the dose to 50 mg twice daily (bid) for the following week. Other rapid-acting NSAIDs like ibuprofen and diclofenac may do just as well, but drugs with long half-lives, such as piroxicam and oxaprozin, should be avoided, since their onset of action is too slow. Always use the highest recommended dose of an NSAID initially, since both pain and inflammation need to be treated promptly.
A cyclooxygenase (COX)-2 selective inhibitor may also be effective. Though etoricoxib has been found to be just as effective as indomethacin in acute gout with much better toleration, it's not currently available in the United States. The potentially adverse cardiac effects of NSAIDs and COX-2 inhibitors are not a concern if treatment is confined to 1 to 2 weeks. Adding a proton-pump inhibitor (PPI) may be prudent in patients with a history of peptic ulcer disease or gastrointestinal (GI) intolerance of NSAIDs.
If NSAID use is contraindicated or not tolerated, what alternatives are available?
Colcrys (colchicine tablets) in 0.6 mg dosage is effective in most acute gout cases. The typical dosage is 1.2 mg followed by a single dose of 0.6 mg 1 hour later for a maximum dose totaling 1.8 mg in the first 24 hours. Colchicine is continued at 0.6 mg daily for up to 3 days, though it should be used at reduced dosage and with caution in patients with renal impairment (CrCl <45 ml/min.).
Gout also responds promptly to intra-articular or systemic corticosteroids. I will occasionally inject a gouty joint with triamcinolone or methylprednisolone acetate followed by ice with good results. Prednisone given orally or methylprednisolone given intravenously are fast-acting and effective, and the IV route may be used in hospitalized patients who cannot be given oral medications. Despite concerns in older literature about rebound gout in patients who are given corticosteroids, recent experience indicates that this does not happen. Patients with moderate-to-severe renal insufficiency are best treated with corticosteroids since NSAIDs may worsen renal failure. Urate-lowering therapy should not be started during an acute attack, since it has no effect on pain or inflammation.
The patient returns 2 weeks later and reports that his pain responded rapidly to indomethacin. What follow-up steps would you recommend?
In patients with a first gouty attack, it is reasonable to limit treatment to resolving the acute attack, to focus attention on comorbid conditions, and to try to lower uric acid levels by diet alone. A weight loss of more than 10 lbs has been shown to lower the risk of gout, and this may be effective if a patient is overweight. A strict, purine-restricted diet has long been advocated to prevent the recurrence of gout. This consists of avoiding meat, poultry, seafood, and internal organs such as liver, kidney, and sweetbreads; meat and yeast extracts; vegetables such as peas, beans, spinach, lentils, and asparagus; beer and beer products; and distilled spirits.
However, this traditional emphasis on a low purine diet may be misplaced, since a recent study showed that moderate intake of purine-rich vegetables didn't increase the risk of gout. Furthermore, a low purine diet is unpalatable to most people, and long-term compliance is dismal. Such a diet lowers serum urate concentrations by only 1 mg/dL, and while a patient with a serum urate level of 9.5 mg/dL has an 80% risk of a recurrent attack in the next 12 months, the risk is still substantial when the level is lowered to 8.5 mg/dL.
Despite such considerations, I routinely recommend that patients eat less red meat and fish, as well as switch to low-fat dairy products as the preferred source of dietary protein. A recent study showed that restricting calories, reducing saturated fat intake, and replacing refined carbohydrates with complex carbohydrates can reduce serum urate levels by 1.7 mg/dL, as well as gouty attacks. Drinking cherry juice and taking 500 mg vitamin C daily also lowers serum urate. On the other hand, drinking 2 or more beers a day increases the risk of another gout attack by a factor of 2.5, so patients should be advised to restrict their intake of beer and distilled spirits. Though 2 glasses of wine a day isn't associated with increased risk, most sweetened, non-alcoholic beverages contain high fructose corn syrup and should be avoided since fructose increases the conversion of ATP to AMP and raises uric acid. However, diet drinks are fine.
Diuretics may modestly increase the serum urate levels — as they did in our patient — and using an alternative antihypertensive agent may lower the risk of a recurrent gouty attack. The angiotensin II receptor blocker losartan potassium is an effective antihypertensive drug that has a uricosuric effect, so it is uniquely beneficial in this setting.
When would you start urate-lowering therapy?
A patient with 2 to 3 recurrent attacks a year should be treated, but if the attacks are severe or disabling, then starting treatment even after the first attack may be reasonable, especially if serum urate is too high to be effectively lowered by diet and lifestyle modification(>9 mg/dL). The presence of tophi, radiographic evidence of erosions, joint space narrowing or deformity, high 24-hour urine uric acid excretion (≥1,100 mg), renal insufficiency, or a history of kidney stones dictate urate-lowering therapy. I find that the presence of gout in the elbows, wrists, or fingers is a sign of chronic recurrent and often tophaceous gout, so I will initiate urate-lowering treatment even if I don't get a clear history of recurrent attacks.
Traditionally, it is recommended that we wait 2 to 4 weeks before starting a urate- lowering drug out of fear that it may trigger another attack or prolong the attack. A recent small study of 51 patients given either placebo or allopurinol at the time of gout attack did not show worsening of gout flare with allopurinol. The advantage of waiting 2 to 4 weeks before starting treatment is that the uric acid level obtained when the patient is not symptomatic more accurately reflects the true height of uric acid elevation. In my experience, the advantage of starting treatment at the time of the attack is that it improves compliance, as a patient frequently will not show up for the return visit when the gout attack is resolved.
Nevertheless, I don't favor starting a urate-lowering agent until the acute gout attack is over. It's advisable to wait about a month after the attack to start treatment.
How would you choose among the agents available?
The goal of uric-acid-lowering therapy is to get the serum urate concentration below the saturation point (6.8 mg/dL). I aim to get the level below 6 mg/dL in all patients and below 5 mg/dL in those with extensive tophi. A study of Japanese patients showed that lowering mean serum urate levels to below 6 mg/dL eliminated recurrent attacks in 86% of patients after 1 year of therapy, and these results were maintained during 3 years of treatment.
I often start with probenecid 250 mg bid for a month and then increase the dosage to 500 mg to 1,000 mg bid over the next 2 months to a maximum of 1,500 mg bid. However, the uricosuric agent is not effective in patients with impaired renal function with a creatinine clearance below 35 mL/min. Side effects including rash and GI intolerance are mild and infrequent.
The biggest potential problem with probenecid is precipitation of uric acid stones in patients with hyperexcretion of uric acid. In light of this, I routinely obtain a baseline 24-hour urinary uric acid and creatinine clearance level prior to starting a course of probenecid. I use Zyloprim (allopurinol) instead of probenecid if creatinine clearance is lower than 50 mL/min or if urinary uric acid excretion exceeds 1,000 mg in 24 hours.
The probenecid dose can be reduced after a year of treatment if serum urate levels remain within target range. Patients who take a cardioprotective dose of aspirin between 81 mg and 325 mg daily do not need to stop the aspirin when they start probenecid, since the drug’s effect on serum urate at that dose is minimal. However, higher doses of aspirin block the uricosuric efficacy of probenecid, so those patients should be taking allopurinol. The risk of precipitating uric acid stones is reduced by drinking at least 2 liters of fluid daily.
The 24-hour urine excretion reveals 1,148 mg uric acid. How do you prescribe allopurinol?
Allopurinol is actually more convenient than probenecid in that it is taken once daily and is effective even in renal failure. It inhibits xanthine oxidase and prevents the conversion of hypoxanthine and xanthine to uric acid. Since efficacy does not depend on uric acid excretion, it is effective in virtually all patients with gout. Allopurinol would actually be the drug of choice for all patients if not for its higher risk of serious side effects. Rash, diarrhea, drug fever, leukopenia, or thrombocytopenia occurs in about 5% of patients, though some successfully stay with allopurinol despite those mild side effects.
The dreaded allopurinol hypersensitivity syndrome is fortunately rare, but it has a mortality rate of 25%. These patients develop a severe erythematous rash, fever, hepatitis, eosinophilia, and acute renal failure. It is reported to occur in those with mild renal insufficiency who are given standard doses of allopurinol together with a diuretic. Recently, the specific genotype HLA-B*5801 was linked to all cases of the allopurinol hypersensitivity syndrome in certain populations. Therefore, genotyping for this allele should be considered in patients of Korean descent who have stage 3 or worse chronic kidney disease, as well as in those of Han Chinese or Thai origin irrespective of renal function. Allopurinol should not be used in patients who test positive for the genotype.
It is essential to limit allopurinol to the minimum dose needed to achieve the desired antihyperuricemic effect. Allopurinol converts to its major active metabolite, oxypurinol, with a half-life of 24 hours, which is prolonged in renal failure and necessitates a dose reduction. I start allopurinol at 100 mg daily and slowly increase the dose 2 to 4 weeks in order to achieve the target serum urate of under 6 mg/dL. The maximum dose of allopurinol may be as high as 800 mg daily. A recent study showed that if the highest dose used was 300 mg a day, then only 9% of patients with baseline urate levels of 10 mg/dL or higher achieve a urate level below 6 mg/dL. A common problem in patients referred to me is that their daily dose of allopurinol was too low to control their gout attacks.
Once a patient reaches the target serum urate level, I monitor serum urate twice a year, mainly to ensure compliance. Almost all patients are free from further gout attacks at 6 mg/dL.
Three weeks into treatment, the patient develops a maculopapular rash. How would you manage this side effect?
Although patients who develop a rash on allopurinol may benefit from a desensitization regimen, I no longer need to perform allopurinol desensitization. Instead, I would switch the patient to febuxostat, an alternative xanthine oxidase inhibitor to allopurinol that is now available.
Febuxostat is a thiazolecarboxylic acid derivative that, unlike allopurinol, is not a purine base analogue. Patients who are allergic to either agent can be safely treated with the other agent without experiencing an increased risk of serious adverse events.
Febuxostat is administered daily as a single oral dose of 40 mg or 80 mg. I typically start with 40 mg daily and obtain a serum uric acid level at 2 weeks of treatment. If it achieves the target level of ≤6 mg/dL, then I stay on that dosage, but if it does not, then I raise it to 80 mg daily. A daily dose of 40 mg is roughly equivalent to the efficacy of 300 mg daily of allopurinol. About half of my patients require the higher dosage. A dosage of 120 mg daily has been approved in Europe, but not in the United States. Since 97% of the drug is metabolized by the liver, no dosage adjustment is needed for patients with reduced renal function. However, patients with severe CKD (CrCl <30) were not studied in clinical trials, so caution should be exercised in these patients.
In clinical trials, patients on febuxostat had a higher incidence of nausea, arthralgia, rash, and liver function tests abnormalities than placebo, but the side effects were similar to allopurinol-treated patients. Febuxostat-treated patients had a higher incidence of cardiovascular events than allopurinol, but this was not seen in a larger randomized trial and in post-marketing data.
Febuxostat should not be used together with allopurinol because there is no additive benefit, but either drug can be used with probenecid for an added urate-lowering effect.
Should we always prescribe anti-inflammatory prophylaxis when we start a urate-lowering agent?
It is well-known that gouty attacks actually increase in frequency at the start of therapy. This is upsetting to the patient, who then considers the treatment plan a failure. To prevent these recurrent attacks, I always add a prophylactic agent when I start either probenecid or allopurinol treatment.
The best strategy is to prescribe colchicine 0.6 mg bid. Elderly patients and those with renal impairment are best treated with colchicine 0.6 mg daily or every other day. Colchicine is then continued for 6 months beyond the date of reaching a target serum urate level below 6mg/dL, but those with tophi may need to take prophylactic colchicine for a year or longer until the tophi are resolved. Slowly increasing the dose of probenecid or allopurinol also reduces the frequency of recurrent gout attacks.
Patients who cannot tolerate colchicine may be treated with a modest dose of an NSAID for prophylaxis. I commonly use naproxen 250 mg bid; ibuprofen 600 mg bid; diclofenac 50 mg bid; or indomethacin 25 mg bid. Patients with renal failure pose a problem since neither colchicine nor an NSAID is safe in that setting. In such a setting, I will generally forego prophylaxis and use very small incremental doses of the urate-lowering agent to find the lowest dose that will reach the target. Patients who cannot take either colchicine or an NSAID can be given a prescription for a 4 mg Medrol DosePak (methylprednisolone) to take at the first sign of a gout flare. This is effective in aborting the attack.
If required to achieve the target uric acid level, can you combine therapies?
Combining allopurinol and probenecid may achieve lower serum urate levels than either agent alone. I use such combination therapy rarely, but when I do use it, it’s usually for severe tophaceous gout. I have found combination therapy to be safe and well-tolerated with more rapid and complete resolution of tophi.
Are there any other options available?
Pegloticase is a porcine uricase linked to methoxy-polyethylene glycol that is an alternative therapy for patients with severe gout in whom treatment with other urate-lowering agents has failed to be effective.
I use pegloticase as an option for patients whose gout is advanced and actively symptomatic, or when the use of other urate-lowering therapies is contraindicated. It consists of a series of IV preparations of polyethylene glycol (PEG) uricase, which is the enzyme that catalyzes the conversion of relatively insoluble urate to highly soluble allantoin. This drug is dramatically effective in reducing the size of tophi in patients with extensive tophaceous disease within 3 months. It may play an important role in treating patients who fail to respond or are allergic to both allopurinol and febuxostat, as well as in organ transplant recipients who are taking cyclosporine.
Patients may develop anaphylactic reactions with this treatment, and these events are heralded by a failure of serum urate to drop below 6 mg/dL after an infusion. Allopurinol and febuxostat should not be given together with pegloticase because they will eliminate this safety signal.
Under what circumstances would you treat asymptomatic hyperuricemia?
Elevated uric acid is considered an independent risk factor for worsening of the disease and poorer prognosis in patients with diabetes, insulin-resistance, hypertension, hyperlipidemia, myocardial infarction, and stroke. However, there is no evidence that lowering uric acid will improve these conditions. Though there is some emerging evidence that lowering uric acid below 6 mg/dL may slow down the progression of CKD, the current consensus is that asymptomatic hyperuricemia — or elevated uric acid with the absence of gout — should not be treated. However, the presence of uric acid stones and the height of hyperuricemia deserve consideration.
Expert opinion suggests that we should consider lowering uric acid in patients with asymptomatic hyperuricemia when serum urate is greater than 13 mg/dL in men or greater than 10 mg/dL in women, since such high values may have some nephrotoxic risk. Furthermore, the risk of such patients developing gout in the near future is high. Excretion of uric acid greater than 1,100 mg in 24 hours confers a risk of developing uric acid renal stones in 50% of patients. Therefore, prophylaxis with allopurinol to reduce levels to <800 mg is reasonable.
Patients who are about to receive either radiotherapy or chemotherapy that is likely to result in extensive tumor cytolysis should be treated to prevent acute uric acid nephropathy and other manifestations of tumor lysis syndrome. Preventive therapy in patients at risk includes intravenous hydration and either allopurinol or Elitek (rasburicase) — a recombinant urate oxidase that resembles pegloticase.
About the Author
Peng Thim Fan, MD, is Clinical Professor of Medicine in the Division of Rheumatology at the David Geffen School of Medicine at UCLA. All questions were posed by Family Practice Recertification Editor-in-Chief Martin Quan, MD.














